Strains and plasmids
A complete list of strains and plasmids used in this study is provided in Supplementary Table 1, and oligonucleotide sequences and strain construction methods are provided in Supplementary Note. Plasmids were generated by Gibson ligation using PrimeSTAR HS DNA polymerase (Takara, R044A) for PCR and Gibson Assembly Master Mix (New England Biolabs (NEB), E2611). Chromosomal engineering was performed by lambda Red integration64. Previously described strains and plasmids are referenced where appropriate in Supplementary Table 1. All engineered DNA regions were verified by sequencing. Oligonucleotides were obtained from Integrated DNA Technologies (IDT) unless otherwise noted. Strains and plasmids generated in this study are available from the corresponding author.
Strain growth conditions
For all gene expression measurements, cultures were prepared in two steps. First, colonies were inoculated in a liquid medium and grown for 8–9 h and diluted >104-fold in fresh medium (same type as the initial culture) for overnight growth. Experiments were performed during exponential growth (optical density (OD)600 = 0.2 for E. coli and B. subtilis; OD660 = 0.2 for C. crescentus).
Unless otherwise noted, E. coli cells were grown in 20–30 ml M9 minimal medium (6 g l−1 Na2HPO4, 3 g l−1 KH2PO4, 0.5 g l−1 NaCl, 1 g l−1 NH4Cl, 2 mM Mg2SO4, 0.1 mM CaCl2) supplemented with 0.2% glycerol, 0.1% casamino acids and 1 mg l−1 thiamine at 30 °C using a water bath shaker. Under these conditions, most strains had doubling times similar to that of the wild type (95.0 ± 9.40 min), except RNase E ΔMTS (117.0 ± 27.0 min).
For the temperature-sensitive RNase E mutant (rne3071), cultures were grown as above until OD600 = 0.2 and shifted to 43.5 °C for 10 min before induction65.
B. subtilis cells were grown in MOPS minimal medium with maltose (1× MOPS mixture, 1.32 mM K2HPO4, 0.2% glutamate, 0.1 g l−1 tryptophan, 0.4% maltose) at 30 °C (doubling time 92 ± 9.22 min).
C. crescentus cells were grown in M2G medium (0.87 g l−1 Na2HPO4, 0.54 g l−1 KH2PO4, 0.50 g l−1 NH4Cl, 0.5 mM MgSO4, 0.5 mM CaCl2, 0.01 mM FeSO4, 0.2% glucose) at 28 °C (doubling time 142 ± 3.33 min).
Induction and re-repression of lacZ expression
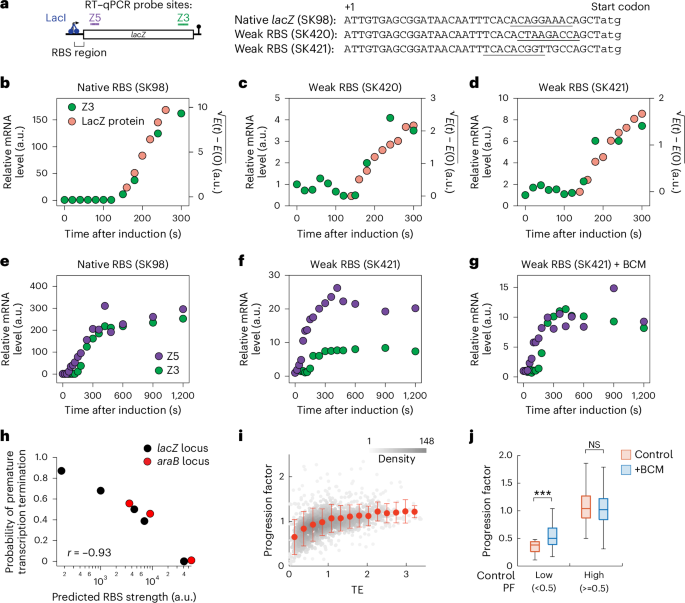
For all lacZ expression under Plac in E. coli, IPTG (0.2 mM) was added at t = 0. To re-repress lacZ expression, 500 mM glucose was added at t = 75 s (ref. 34). When bicyclomycin (BCM) was used, 100 µg ml−1 BCM was added 5 min before IPTG addition.
The glucose addition time was chosen on the basis of two factors: First, it should be more than 1 min before T3′ so that the time window i is sufficiently long for kd1 fitting. Since we withdrew samples every 15–20 s, ~3–4 samples could be taken during the time window i of 1 min for kd1 estimation. However, if glucose was added too early, not much Z5 signal developed. Considering these two factors, we decided to add glucose at t = 75 s for lacZ expression in our experimental condition. This glucose addition time was varied for a different gene (araB) and under different growth conditions because T3′ (transcription elongation time) changed. For example, glucose was added at t = 50 s for rne3071 at 43.5 °C. For genes under Para (lacZ and araB), expression was induced with 0.2% arabinose at t = 0 and repressed with 500 mM glucose at t = 75 s for lacZ and t = 60 s for araB. Repression occurs through cAMP receptor protein-dependent regulation of the Para promoter.
For lacZ expression in B. subtilis, 5 mM IPTG was added at t = 0 and transcription initiation was inhibited with 200 μg ml−1 rifampicin at t = 30 s. This rifampicin addition time was chosen on the basis of the same considerations used for glucose addition time.
For lacZ expression in C. crescentus, induction was performed with 0.3% D-xylose at t = 0, and transcription initiation was blocked with 200 μg ml−1 rifampicin at t = 50 s. When BCM was used, 100 µg ml−1 BCM was added 5 min before induction.
RNA extraction and RT–qPCR
At each time point, 0.4 ml of culture was withdrawn and immediately mixed with an equal volume of pre-cooled RNAlater Stabilization Solution (Life Technologies). Samples were incubated on ice for 20 min and centrifuged. Pellets were resuspended in lysozyme solution (10 mg ml−1 lysozyme in 10 mM Tris-HCl (pH 8) with 1 mM EDTA). Total RNA was extracted using the PureLink RNA mini kit (Life Technologies) with on-column DNase treatment. RNA was eluted in 50 µl RNase-free water. RNA concentrations were typically 60–80 ng µl−1 (measured using a NanoDrop One Microvolume UV–Vis spectrophotometer). RT–qPCR was performed using KAPA SYBR FAST qPCR Master Mix (KAPA Biosystems) and a Bio-Rad CFX Connect system. Primer sequences are provided in Supplementary Table 2.
For very weak RBS strains (SK626 and SK637) and selected genes analysed in Fig. 4, Taqman probes were used. Complementary (c)DNA synthesis was performed using SuperScript III (Invitrogen), followed by RT–qPCR using TaqMan Universal Master Mix II with UNG (Applied Biosystems).
Analysis of time-course RT–qPCR data
Relative RNA levels were calculated as fold change relative to baseline (pre-induction samples) using the \(^_}}}\) method. Here, Ct denotes the cycle threshold at which the signal crosses the detection threshold.
T5′ and T3′ were obtained by linear fitting of the initial increase in the 5′ and 3′ signals and finding their intercept with the baseline. PT was calculated from the ratio of the initial 3′ slope to the initial 5′ slope under no-glucose experiments.
RNA degradation rates were obtained by fitting log-transformed RNA levels within defined time windows. For example, for data shown in Fig. 2c, Z5 values between t = 150 and 210 s were used to calculate kd1, and Z5 values between t = 300 and 600 s were used to calculate kd2. Z3 values between t = 300 and 600 s could be used to calculate kd2 of Z3, but we focused our analysis on the degradation of Z5. These time windows were adjusted depending on transcription elongation time, which varies with gene length, RNAP speed and glucose addition time34. Phenomenologically, t5′, T3′ and t3′ were identified from the time when Z5 reached maximum, when Z3 started to appear from the baseline and when Z3 reached maximum, respectively. All raw RT–qPCR data are provided in Source Data 1 and fitting results in Source Data 2.
Miller assay
LacZ activity was measured using a modified Miller assay34. At each sampling time, 900 µl of cell culture was withdrawn and immediately mixed with a pre-cooled stop solution. The stop solution was 100 µl 5 mg ml−1 chloramphenicol for E. coli and C. crescentus, and 100 µl 10 mg ml−1 chloramphenicol and 10 mg ml−1 erythromycin for B. subtilis. Cells were washed by centrifugation at 7,000 × g for 1 min (E. coli), 18,200 × g for 4 min (B. subtilis) or 6,000 × g for 4 min (C. crescentus) at 4 °C, and cell pellets were resuspended in 340 µl of 1× Z buffer (30 mM Na2HPO4, 10 mM NaH2PO4, 5 mM KCl, 0.5 mM MgSO4) containing the same concentration of chloramphenicol (and erythromycin) as before the wash. Cells were lysed using SDS and β-mercaptoethanol.
For standard measurements, ortho-nitrophenyl-β-galactoside (ONPG) was used as the substrate, and absorbance was measured at OD420 in a microplate reader (Synergy HTX multimode reader, BioTek). LacZ activity was calculated using standard Miller assay formulas based on OD420, OD550 and OD600. For weak-RBS strains (SK421, SK626 and SK637) and non-E. coli species, the fluorogenic substrate MUG was used. In this case, LacZ activity was calculated from F350/460 and OD600, measured from the plate reader.
Baseline activity was calculated from samples before LacZ induction and subtracted from all timepoints. Translation time (when the first LacZ proteins appeared) was determined by fitting the square root of LacZ activity to the initial linear increase66.
When lacZ expression was re-repressed at t = 75 s, LacZ protein levels reached a plateau at ~t = 7 min. LacZ protein activity at the plateau was calculated by averaging data points between t = 7 and 10 min, and the difference from the baseline was used as a proxy for the total LacZ protein produced during the 75-s induction. When comparing LacZ protein levels across strains, we converted MUG-based LacZ measurements to the ONPG-based scale using the expression levels of SK98 and SK421 measured by MUG as an internal conversion control.
FISH microscopy
E. coli lacZ FISH was performed as described previously34,40. For hybridization, we used 24 single-stranded DNA probes complementary to the first 1-kb region of lacZ mRNA (Z5FISH). Each probe was labelled with a single Cy3B at the 5′ end. Probe sequences are listed in Supplementary Table 3.
Phase-contrast and fluorescence images were taken in the Eclipse Ti-2 microscope (Nikon) equipped with Sola SE II 365 light engine (Lumencor), a phase-contrast objective Plan Apochromat (×100/1.45 NA, Nikon) and an Orca-R2 CCD camera (Hamamatsu Photonics). Cell outlines were detected using Oufti67, and fluorescent spots were identified using u-track68 with Gaussian mixture-model fitting (α = 0.015 for local maxima detection; α = 0.05 for Gaussian peak testing). Custom MATLAB scripts were used to assign spots to individual cells and calculate spot number and localization (see Code availability). Spot positions were normalized to cell dimensions and visualized as 2D histograms. Number of cells and detected spots are summarized in Supplementary Table 4.
TE and PF analysis
Translation efficiency (TE) values were obtained from published ribosome profiling data (Supplementary Table 4 in ref. 25). Transcription progression factor (PF) values were calculated from published SEnd-seq data28 (GEO access number GSE117737). For each protein-coding gene, upstream (from 0 to 200 nt downstream of the TSS) and downstream (from 500 nt downstream of the TSS to the end of the gene) zones were defined, and the ratio of downstream to upstream signal was used to calculate PF. If there was another qualified TSS located within the downstream zone, the region downstream of that TSS was excluded from analysis. Genes with a short length (<600 bp) or a low expression level (<50 SEnd-seq reads) were removed from the analysis. TE and PF data are summarized in Source Data 3.
Rif-seqSample collection and RNA purification
E. coli wild-type cells (MG1655) were grown in MOPS EZ rich defined medium (Teknova, M2105) at 37 °C. An overnight culture was diluted into fresh medium to an initial OD600 of 0.005 and grown to mid-log phase (OD600 ≈ 0.3). Rifampicin (Sigma-Aldrich, R3501) was added at a final concentration of 500 µg ml−1 to inhibit transcription initiation. Samples were collected at t = 0, 0.5, 1, 2, 3, 4, 5, 6, 8 and 10 min after rifampicin treatment by mixing 2 ml of culture with 250 µl of ice-cold stop solution (225 µl ethanol and 25 µl phenol). Cultures were centrifuged at 8,200 × g for 3 min, and pellets were flash frozen in liquid nitrogen.
Total RNA was purified from samples using hot acid phenol extraction. Briefly, frozen pellets were resuspended in 500 µl RNA extraction buffer (0.3 M NaOAc, 1 mM EDTA, 1% SDS) and mixed with 600 µl hot phenol (Invitrogen, AM9720) at 65 °C for 5 min. The mixture was centrifuged at 18,400 × g at 4 °C for 10 min, and the upper aqueous layer was transferred to a new tube. A second phenol/chloroform extraction was performed, followed by an additional extraction with an equal volume of chloroform. The upper layer was mixed with 2 µl GlycoBlue Coprecipitant (Invitrogen, AM9516) and an equal volume of isopropanol (Sigma-Aldrich, I9516) to precipitate RNA. RNA pellets were washed with 750 µl 80% ethanol, dried and resuspended in 10 mM Tris pH 7. Genomic DNA was removed using Turbo DNase (Invitrogen, AM2238).
RNA-seq library construction
Purified RNA was first fragmented by mixing with an equal volume of 2× alkaline fragmentation buffer (0.6 volumes of 100 mM Na2CO3 and 4.4 volumes of 100 mM NaHCO3), incubated at 95 °C for 13 min and then purified using Zymo oligo CC kit (Zymo Research, D4060). Fragmented RNA (600 ng) was dephosphorylated using T4 polynucleotide kinase (NEB, M0201) at 37 °C for 1 h, followed by ligation to 3′ linker oligos (sequences listed below) using T4 RNA ligase 2 truncated KQ (New England Biolabs, M0373) at 16 °C overnight.
3′ linkers: /5rApp/NNNNNXXXXAGATCGGAAGAGCGTCGTGTAGGGAAAGA/3SpC3/
XXXX (4-base barcode): t = 0 CGAT; t = 0.5 GCTA; t = 1 ATCG; t = 2 TAGC; t = 3 GACT; t = 4 AGTC; t = 5 TCAG; t = 6 CTGA; t = 8 CATG; t = 10 TGAC.
The ligated products from different samples were pooled and purified by size excision on a Novex 10% TBE-urea polyacrylamide gel (Invitrogen, EC68752BOX). The purified pooled ligation products were then split into two tubes: one for total RNA and the other for mRNA enrichment. For samples requiring mRNA enrichment, ribosomal RNAs were removed from the total RNA with Ribo-Zero rRNA depletion kit (Epicentre, MRZGN126). For both total RNA-seq and mRNA-seq libraries, ligated RNA was reverse transcribed into cDNA by SuperScript IV Reverse Transcriptase (Invitrogen, 18090010) (reverse transcription primer 5′-TCTTTCCCTACACGACGCTC-3′). The resulting cDNA was purified using Zymo Oligo CC kit. cDNA fragments were ligated to a second oligo (/5rApp/NNNNNCTGAAGATCGGAAGAGCACACGTCTGAACTC/3ddC/) using T4 RNA ligase 1 (New England Biolabs, M0204) at 16 °C overnight. The final ligation products were purified by size excision on a Novex 10% TBE-urea polyacrylamide gel. Finally, the purified cDNAs were PCR amplified using Phusion polymerase (New England Biolabs, M0530) and the following PCR primers:
Forward primer:
5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTC-3′;
Reverse primer:
5′- CAAGCAGAAGACGGCATACGAGATCTTGTAGTGACTGGAGTTCAGACGTGTGCTC-3′ (of note, the 6-base index sequence CTTGTA can be replaced to allow multiplexing). After 8–10 cycles of PCR amplification, libraries were size selected on a Novex 8% TBE gel (Invitrogen, EC6215BOX) and purified. Sequencing was performed on an Illumina HiSeq 4000 system, using single-end 50-nucleotide reads (Center for Advanced Technology, UCSF).
Sequencing data processing
The raw sequencing reads were first demultiplexed on the basis of the 4-base barcode corresponding to individual samples pooled in the library using fastx_barcode_splitter (https://github.com/agordon/fastx_toolkit). The linker sequence was clipped using Cutadapt69. The clipped sequences were then mapped to the reference genome NC_000913.3, obtained from the NCBI Reference Sequence Database, using STAR (v.2.7.6a)70. The following parameters were used:
–alignEndsType EndToEnd –outSAMmultNmax -1 –outFilterMismatchNmax 2 –clip5pNbase 9 –outSAMtype BAM SortedByCoordinate –outWigType wiggle read1_5p –outWigStrand Stranded –outWigNorm None.
The STAR alignment output files are stranded wiggle files. ‘Signal.UniqueMultiple.str1’ and ‘Signal.UniqueMultiple.str2’ represent signal from both uniquely and multimapped reads mapped to the minus and plus strand, respectively. Multimapping signals were distributed equally across loci (for example, for ribosomal RNA reads).
Rif-seq data analysis
mRNA levels of individual genes before and after Rif treatment were calculated as follows. Since the overall levels of unstable RNAs decrease after Rif treatment due to degradation, we used a normalization standard to account for this change between timepoints. When E. coli is grown under optimal growth conditions, the majority of total RNA consists of ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs), which are generally considered stable due to their significantly longer half-lives compared with mRNA. Using the total RNA-seq dataset, we divided all sequencing reads into two groups: stable RNAs, including rRNAs and tRNAs; and unstable RNAs, including mRNAs (mapped to protein-coding genes) and other non-coding RNAs (ncRNA). For each time point, we calculated the ratio of unstable RNA reads (mRNAs and ncRNAs) relative to the total stable RNA reads (rRNAs and tRNAs) as a normalization standard, referred to as ‘unstable RNA%’.
From total RNA-seq at each time point:
$$}\, } \% =\frac}\,}\,}\,}\,}\,}}}\,}\,}\,}\,}\,}}$$
(1)
This unstable RNA% value was used to normalize the mRNA-seq reads per kilobase million (RPKM) values and to quantify the abundance of individual mRNAs across timepoints following Rif treatment. The adjusted mRNA RPKM (aRPKM) for gene i at a specific time point was calculated as:
From mRNA-seq at the same time point:
$$\begin}_ &=\frac}\, }\, }\, }\, }\, }\, \times }}}\, \,}\, (}) \times}\, }\, }\,}\, }\,}} \\&\quad \times }\,}\%\end$$
(2)
Calculation of mRNA half-lives
We applied the following filters to select actively expressed protein-coding genes for half-life calculation: (1) the number of mRNA-seq reads mapped to the gene at time point t = 0 was at least 100 and (2) the gene had non-zero mRNA-seq read counts at all subsequent timepoints. A total of 996 protein-coding genes passed these filters.
If mRNAs decay exponentially, their half-lives can be calculated as follows:
$$R\left(t\right)=_\times ^};_=\mathrm(2)/k$$
(3)
where R(t) is the RNA abundance at time t, R0 is the RNA abundance at time 0 (before Rif treatment), k is the degradation rate constant, and T1/2 is the RNA half-life.
Therefore, for each gene that passed the filters, we first normalized mRNA levels at different timepoints to the level at t = 0. We then applied a log transformation to these normalized values and fitted them to a linear model across the timepoints. To capture both early and late phases of mRNA decay, we performed piece-wise linear regression using the R package ‘segmented’, which can be found at https://www.rdocumentation.org/packages/segmented/versions/2.1-2. We used the ‘davies.test’ function to test for a change in slope. If the adjusted P value was lower than 0.05 (that is, two slopes are significantly different), the two separate half-lives along with the corresponding time breakpoint were calculated. Otherwise, the data were fitted into a single linear model, and the single half-life was calculated. The raw data and processed data are provided in Source Data 4.
RT–qPCR validation of Rif-seq decay profiles
To validate two-phase decay patterns observed in Rif-seq, decay kinetics were measured by RT–qPCR after rifampicin treatment. Cells (SK98 or SK626) were grown in MOPS EZ rich defined medium at 37 °C until OD600 = 0.1 and IPTG (0.2 mM) was added at t = −20 min. Rifampicin (500 µg ml−1) was added at t = 0, and samples were collected over 10 min. When BCM was used, 100 µg ml−1 BCM was added at t = −10 min. RNA extraction and RT–qPCR were performed as described above using Taqman probes.
Simulation model for Rif-seq and two-phase decay pattern
To investigate the origin of the two-phase decay pattern observed in our Rif-seq data, we performed stochastic simulations of transcription and mRNA degradation using experimentally derived parameters. The simulations included RNAP initiation, elongation, premature termination and mRNA decay, and were carried out using an extended version of a previously developed totally asymmetric simple exclusion process (TASEP)-based framework71, in which premature termination was newly incorporated. In the model, we assumed that premature termination takes place in the middle of a 3,075-bp DNA template at a certain percentage (input parameter, PT). We had the first base of the mRNA (5′ end) decay either at kd1* (during transcription elongation, as nascent mRNA), at kdPT (if prematurely released) or at kd2 (if transcription was completed at the gene end). This initial degradation was followed by 5′-to-3′ directional degradation. At 0.1-s intervals along the simulation, the copy number of 5′ mRNA (or any other positions) was counted. The mean copy number was calculated from 300 iterations of the simulation.
Similar to lacZ induction experiments, the simulated mRNA level increased from zero and reached a steady state before t = 10 min. Transcription initiation was halted (similar to Rif-seq) at t = 20 min of simulation time. For presentation, we plotted the log mRNA levels after transcription inhibition (re-defined this time as t = 0). Since the steady-state levels were different depending on the input parameters, log mRNA levels were shifted so that they start from 2 at t = 0 (Fig. 4d and Extended Data Fig. 8h,i).
Statistical test
All replicates represent biological replicates. Two-sample unpaired Student’s t-tests were performed using MATLAB (ttest2 function). P > 0.05 is marked as NS for not significant. Statistical test results are summarized in Supplementary Table 5.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.


Comments (0)