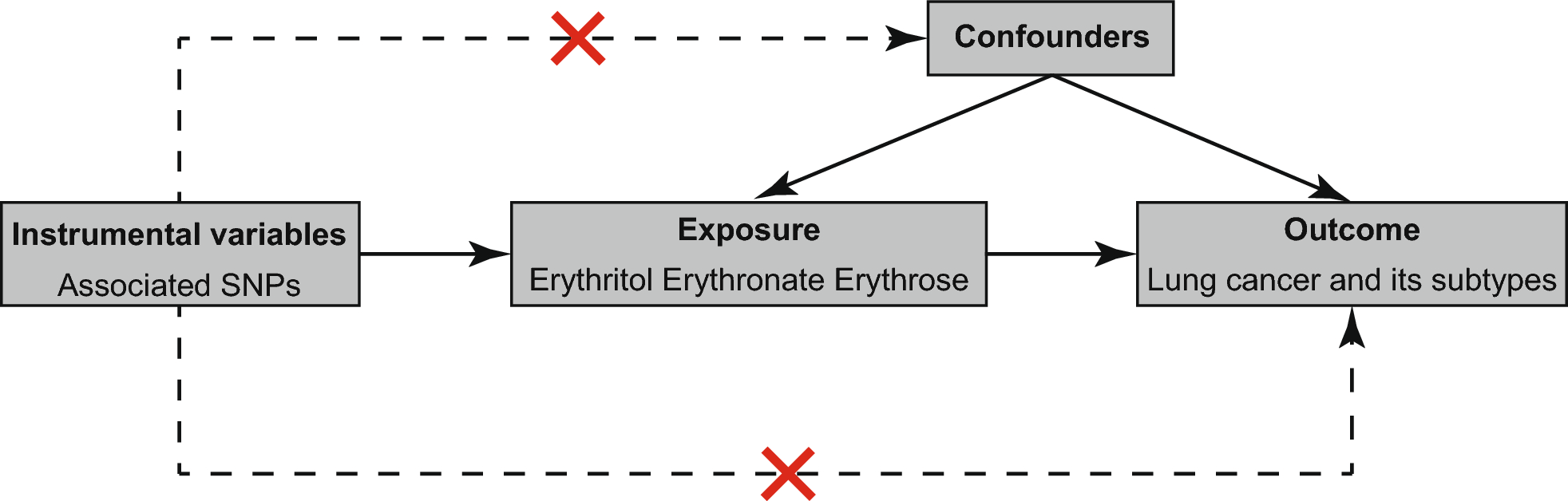
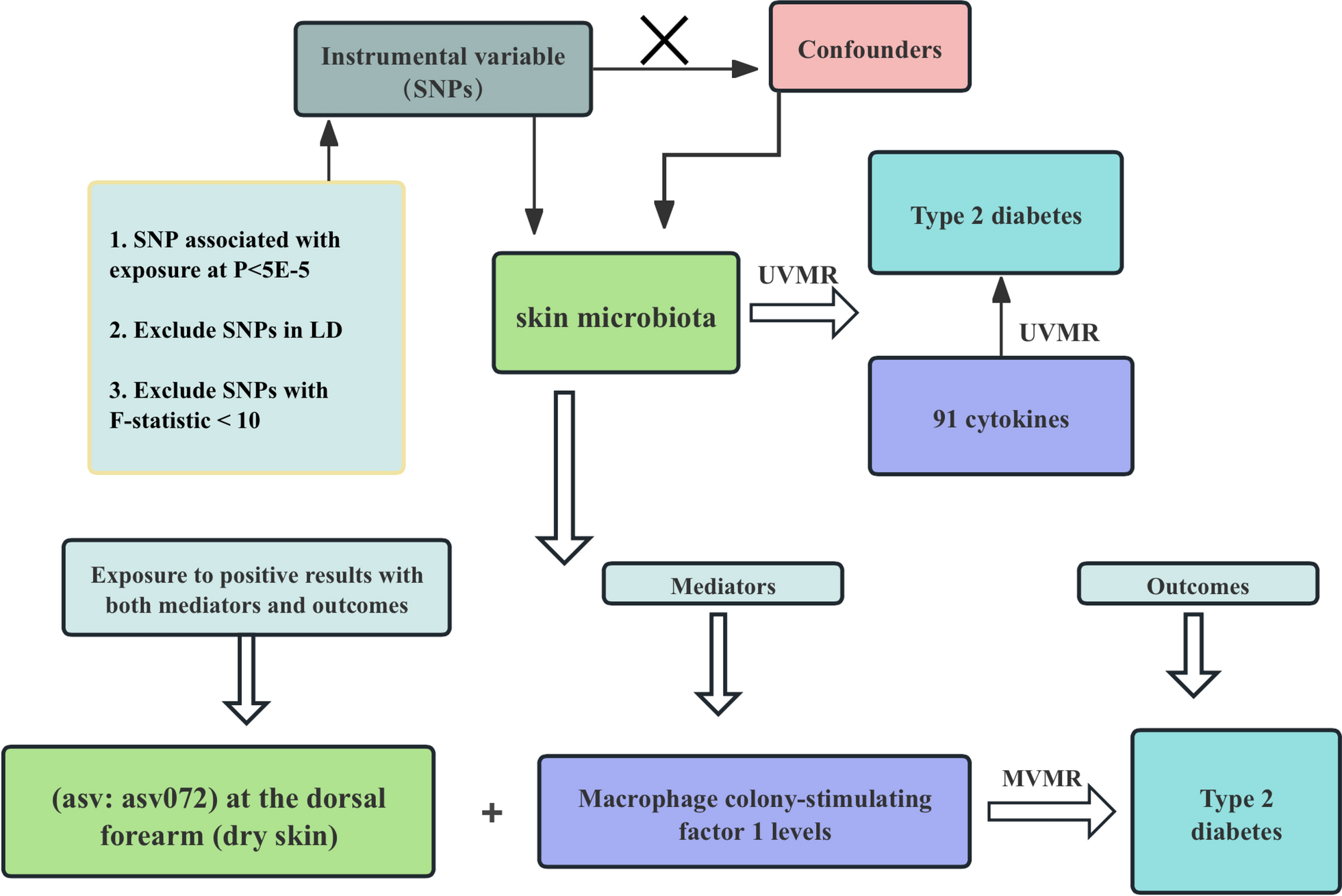
In this study, we delve deeply into the rich GWAS meta-analysis data resources from Biobank PopGen and KORA Genzentrum, aiming to uncover the intricate associations between skin microbiota and T2DM, and to explore potential mediating mechanisms. The research findings indicate that a series of specific microbial communities are closely related to T2DM risk: genus: Acinetobacter, class: Alphaproteobacteria, ASV005 [P. granulosum], ASV072 [R. mucilaginosa], and genus: Bacteroides were all identified as factors increasing the risk of T2DM. Conversely, phylum: Proteobacteria, genus: Enhydrobacter, family: Clostridiales and ASV006[Staphylococcushominis] demonstrated a protective effect against T2DM. Notably, we discovered that the association between ASV072 [R. mucilaginosa] and T2DM risk is not isolated but indirectly affects the risk of T2DM through the mediating pathway of regulating Macrophage colony-stimulating factor 1 (M-CSF-1) levels. This finding not only broadens our understanding of the role of skin microbiota in the pathogenesis of T2DM but also provides a strong scientific basis for future development of T2DM prevention and treatment strategies based on microbial community modulation.
Previous studies on the microbiota and T2DM have largely focused on the gut microbiota. Gut microbiota dysbiosis has been shown to impair gut barrier function, leading to systemic inflammation and the development of insulin resistance. In particular, endotoxins such as lipopolysaccharides (LPS) produced by gut bacteria can trigger chronic low-grade inflammation, which negatively impacts insulin action [27]. Furthermore, gut microbes ferment dietary fibers to produce short-chain fatty acids (SCFAs) like acetate and butyrate, which play a crucial role in regulating systemic metabolism, insulin sensitivity, and fat storage. Dysbiosis may reduce SCFA production, thereby increasing the risk of T2DM [28]. Additionally, gut microbiota affects fat metabolism by modulating the synthesis, breakdown, and storage of fatty acids, influencing insulin sensitivity and glucose metabolism. Imbalances in gut microbiota can disrupt these processes, promoting T2DM. The gut microbiota also regulates bile acid metabolism, which plays a key role in energy and glucose metabolism, with dysbiosis potentially leading to altered bile acid metabolism and further promoting T2DM [29]. Moreover, gut microbiota influences insulin secretion, particularly through the modulation of gut-derived incretin hormones like GLP-1, which are crucial for insulin release. Dysbiosis may reduce GLP-1 secretion, impairing insulin sensitivity and glucose metabolism [30]. Lastly, interactions between gut microbes and host gene expression can alter the activity of enzymes involved in glucose metabolism, further affecting insulin action and glucose homeostasis [31].
Existing studies have confirmed that the composition of the microbiome changes in different biological environments during alterations in the host immune system or other pathological states, and patients with T2DM exhibit dysbiosis of skin microbial communities [32], possibly stemming from the same activation of innate immune responses. When lesions occur, skin microbes can invade affected tissues and stimulate inflammatory cascade reactions by providing pathogen-associated molecular patterns (PAMP) signals, thereby triggering local immune responses. Therefore, the skin microbiota may increase the risk of diabetic skin signs and even promote the occurrence of Diabetic Foot Wounds (DFW) [33]. Recent studies have shown that skin stressors (such as dryness and barrier disruption) can stimulate the production of cortisol in the skin, an effect that has systemic impacts through the activation of inflammatory cytokines (such as IL-1b) [34]. Dysbiosis of the skin microbiota can lead to skin inflammation and trigger a large infiltration of immune cells, causing barrier disruption and local skin damage. Once inflammatory mediators from the skin enter the systemic circulation, the release of pro-inflammatory cytokines can induce chronic systemic inflammation and metabolic syndrome, which may be another mechanism linking the skin microbiome to diabetes [35]. Additionally, it has been reported that the analysis of the diabetic skin microbiota can not only serve as a potential diagnostic marker for targeted antibiotic treatment but also aid in developing new therapeutic strategies, such as probiotics or probiotic formulations, which can regulate pathogenic skin flora and form new bacterial communities. By using specific antibiotics, these flora can be almost completely eradicated and significantly reduce skin inflammation [36, 37]. Therefore, more research is needed on the association between skin microbiota and T2DM.
Research has posited that inflammatory responses may constitute the pivotal nexus between the skin microbiota and the onset of T2DM. Acinetobacter baumannii, a Gram-negative coccobacillus of the Acinetobacter genus, is an obligate aerobe and non-lactose-fermenting conditional pathogen. Due to its high transmissibility, it has become one of the common pathogens. In a study, Acinetobacter baumannii was detected in the serum of 23% of T2DM patients [38], and these patients had significantly elevated levels of pro-inflammatory cytokines, including IL-1b, TNF-a, MCP-1, IL-6, IL-8, and IFN-g, which were not noticeably increased in T2DM patients not infected with Acinetobacter baumannii. Some of these pro-inflammatory cytokines have been considered potential therapeutic targets for diabetes and related chronic diseases. Further research found that compared to healthy individuals [39, 40], the co-occurrence rate of cytokines was higher in diabetic subjects, especially the association between Acinetobacter baumannii and the neutrophil inflammation signaling cytokine IL-8 was more significant [41]. This suggests that in individuals with diabetes, neutrophils may exhibit a set of gene expression differences that trigger abnormal inflammatory responses.
Alphaproteobacteria is a class under the phylum Proteobacteria, consisting of bacteria with diverse metabolic processes that can either form symbiotic relationships with plants and animals or be pathogenic [42]. The bacteria in Alphaproteobacteria may be closely associated with the development of diabetes through their lipopolysaccharide (LPS) [27]. As Gram-negative bacteria, members of Alphaproteobacteria (such as Rickettsia, Rhizobium, etc.) possess LPS as a major component of their cell walls, which can trigger inflammatory responses by interacting with the host immune system. LPS binds to Toll-like receptor 4 (TLR4) on host cells, activating the NF-κB signaling pathway, leading to the release of pro-inflammatory cytokines (such as TNF-α, IL-6, etc.) [43]. These cytokines are key drivers of insulin resistance and diabetes. The increase in LPS can promote a systemic inflammatory response, inhibiting the action of insulin and thus exacerbating the development of diabetes [43, 44]. Proteobacteria, a diverse bacterial phylum, includes several subgroups that may play a protective role in T2DM. One potential mechanism involves the balance of the gut microbiota [45]. Studies have suggested that an increase in Proteobacteria could work synergistically with beneficial bacterial populations, such as Lactobacilli and Bifidobacteria, to maintain a healthy microbial balance in the gut [46]. This balance may help reduce chronic inflammation and prevent insulin resistance, both of which are critical factors in the development of T2DM. Furthermore, certain members of Proteobacteria have immune-modulatory functions. They can produce SCFAs, such as butyrate, which inhibit inflammatory responses and improve insulin sensitivity, thereby exerting a protective effect against T2DM [47, 48]. Additionally, some Proteobacteria members may enhance gut barrier function, reducing the penetration of harmful bacteria and endotoxins, alleviating endogenous inflammation, and supporting overall metabolic health [49]. Bacteroides, as a Gram-negative, anaerobic bacterium belonging to the family Bacteroidaceae, is widely present in the gut of humans and animals and is a key member of the gut microbial community. Recent academic research has revealed a potential correlation between Bacteroides and T2DM [50]. Research has indicated that individuals categorized as Enterotype Bacteroides exhibit notably elevated levels of serum LPS compared to healthy subjects. This surge in LPS concentrations can give rise to endotoxemia and a subtle inflammatory reaction, ultimately diminishing insulin sensitivity and accelerating the development of T2DM [44, 50].
The pathogenesis of T2DM is complex, with abnormal autoimmune responses in pancreatic β-cells being one of the core factors. When the immune system mistakenly identifies pancreatic β-cells as foreign antigens and launches an attack, it leads to cell damage or even apoptosis, thereby disrupting the normal secretion and function of insulin, ultimately causing elevated blood glucose level [3]. Clostridiales are a class of Gram-positive bacteria belonging to the order Bacillales. According to a study, diabetic individuals exhibited significantly lower relative concentrations of Clostridiales compared to healthy controls [51]. Clostridium bacteria play a key role in maintaining intestinal barrier integrity and regulating host metabolism by producing SCFAs, especially butyrate, which is utilized by enterocytes to maintain gut health [52]. A reduction in Clostridiales has been associated with dysbiosis in diabetic individuals, leading to impaired gut function and potentially contributing to disease progression [51]. Furthermore, an imbalance between Clostridiales and Lactobacillus could further disrupt gut microbiota, exacerbating dysfunction. Additionally, the decrease in Clostridiales has been linked to aging, a known risk factor for diabetes, suggesting that the loss of these bacteria might be a significant factor in the pathogenesis of diabetes, especially in older populations [53, 54]. Overall, Clostridiales appear to play a crucial role in maintaining gut health and microbial balance, which could be protective against the development and progression of diabetes. Propionibacterium granulosum, a resident skin anaerobe, exhibits higher pathogenicity in diabetic wound infections, contrasting sharply with common pathogens in non-diabetic states [55, 56]. This bacterium has significant immunomodulatory capabilities, capable of stimulating immune system responses through specific strains, such as spleen enlargement and tumor metastasis inhibition, which may further affect the body's inflammatory response and autoimmune status. Notably, the combination of P. granulosum and LPS in vitro experiments can significantly promote the release of various cytokines by alveolar macrophages and peripheral blood mononuclear cells (PBMCs), including IL-1, IL-6, IL-12, and IFN-γ. While these cytokines enhance humoral and cellular immune responses, they may also exacerbate the body's inflammatory state, adversely affecting insulin signaling and glucose metabolism, thus becoming a potential risk factor for the development of T2DM [57, 58].
Rothia mucilaginosa is a Gram-positive bacterium belonging to the phylum Actinobacteria, class Actinobacteria, order Micrococcales, and family Micrococcaceae. Although this bacterium is generally considered to have low virulence, increasing evidence suggests that it is an important opportunistic pathogen affecting immunocompromised hosts [59]. Current research indicates that Rothia mucilaginosa is associated with a variety of severe infections, particularly in immunocompromised hosts, such as cancer patients, those with severe neutropenia, human immunodeficiency virus (HIV) infected individuals, chronic liver disease patients, and diabetics [60, 61]. In the MVMR analysis, we proposed M-CSF-1 as a potential mediator of the impact of Rothia mucilaginosa on T2DM. M-CSF-1 plays a critical role in inflammation and insulin resistance, while Rothia mucilaginosa may influence the production of M-CSF-1 through its regulation of immune responses, thereby indirectly affecting the development of T2DM. Although this model assumes that the genetic instruments act on T2DM exclusively through M-CSF-1 without horizontal pleiotropy, and that the temporal relationship between exposure and outcome is consistent with causality, it is important to consider the potential influence of complex immune-microbiota interactions and residual confounding. Further experimental studies are essential to validate this causal pathway and elucidate its underlying mechanisms.
Our investigation further underscores the existence of distinct protective microbial consortia in individuals afflicted with T2DM, among which Enhydrobacter and Staphylococcus hominis are particularly noteworthy. Enhydrobacter, as a unique extremely short rod-shaped bacterium, shows a significant downward trend in its quantity compared to healthy controls in the deep analysis of intestinal microbial communities of diabetic foot patients [62]. Enhydrobacter, as a member of the resident intestinal flora, may be involved in multiple physiological processes such as host energy metabolism regulation, immune response modulation, and inflammation reaction balance. Its reduced quantity may disrupt the intestinal microecological balance, thereby affecting the generation of SCFAs, intestinal mucosal barrier function, and immune system homeostasis, eventually linking to the pathogenesis of T2DM [62]. On the other hand, Staphylococcus hominis, as a member of the Staphylococcus genus and a common human commensal bacterium, especially in individuals with low immune function, is often regarded as an opportunistic pathogen as part of the Coagulase-negative Staphylococci (CoNS). Although S. hominis is usually associated with infections in newborns and immunosuppressed patients, possessing high antibiotic resistance and biofilm-forming capabilities [63, 64], this study identified its potential protective role in T2DM patients. This discovery may be attributed to the biofilm-forming ability of S. hominis, which could form a protective barrier in diabetic patients, limiting the growth of other harmful bacteria. Additionally, its high resistance allows S. hominis to survive and maintain its population during antibiotic treatment, further preventing infections caused by other susceptible bacteria [65, 66]. Notably, if future research can confirm the ability of S. hominis to regulate immune responses in T2DM patients, it might serve as an important protective bacterium, influencing the progression of diabetes by modulating the host's immune response. This finding not only challenges the traditional perception of S. hominis as a potential pathogen but also provides a new perspective and potential targets for microbial treatment strategies for T2DM.
In this MR study, we identified a causal relationship between three inflammatory factors—Tumor Necrosis Factor Receptor Superfamily Member 9 (TNFRSF9), Urokinase-Type Plasminogen Activator (uPA), and C–C Motif Chemokine 28 (CCL28)—and Type 2 Diabetes Mellitus (T2DM), contributing to the growing body of evidence linking chronic inflammation with the pathogenesis of T2DM. TNFRSF9, also known as 4-1BB, is a member of the tumor necrosis factor receptor superfamily that plays a crucial role in regulating immune responses, particularly the activation and expansion of T cells [67]. Dysregulation of TNFRSF9 signaling has been implicated in various autoimmune diseases and inflammatory conditions, including insulin resistance, a key feature of T2DM [68]. Elevated levels of TNFRSF9 are associated with chronic low-grade inflammation, which disrupts insulin signaling and promotes insulin resistance by enhancing the secretion of pro-inflammatory cytokines such as TNF-α and IL-6 [68]. These mechanisms highlight the potential role of TNFRSF9 as a mediator in the development of T2DM. uPA, a serine protease involved in the plasminogen activation system, plays a vital role in extracellular matrix remodeling and tissue repair [69]. However, dysregulation of uPA has been linked to metabolic disorders, including T2DM. uPA can promote inflammation by activating various signaling pathways that increase the production of pro-inflammatory cytokines, contributing to insulin resistance [70]. Furthermore, uPA's role in extracellular matrix breakdown may facilitate immune cell infiltration into adipose tissues, exacerbating inflammation and metabolic dysfunction. Elevated uPA levels have been associated with insulin resistance and obesity, both of which are risk factors for T2DM [70]. These findings suggest that uPA may be a key player in the inflammatory processes that contribute to T2DM. CCL28 is a chemokine involved in immune cell recruitment to inflammatory sites. It is highly expressed in adipose tissue and the pancreas, critical sites in the pathogenesis of T2DM [71]. CCL28 facilitates the recruitment of T cells and macrophages to these tissues, contributing to insulin resistance by promoting local inflammation. Elevated CCL28 levels are associated with increased adiposity and impaired glucose homeostasis, suggesting its involvement in T2DM progression [72]. Additionally, CCL28 interacts with other inflammatory mediators, such as TNF-α and IL-6, amplifying the inflammatory response and further promoting insulin resistance [73]. These findings underscore the importance of CCL28 in mediating the chronic inflammation underlying T2DM.
Although this study is the first to use MR methods to analyze the causal relationship between skin microbial communities and type 2 diabetes (T2DM), achieving significant research outcomes, it still has some limitations. Primarily, although MR methods can effectively reduce the bias of confounding factors, they cannot completely eliminate these biases. The results are still dependent on the validity of the instrumental variables used, and any violations of the assumptions (such as pleiotropy or weak instruments) could affect the robustness of the findings. To mitigate this limitation, we employed various robust MR analysis methods and conducted sensitivity analyses to verify the consistency and reliability of our research results. Secondly, the data used in this study mainly come from individuals of European descent, which may limit the generalizability of the findings to other populations. The broad applicability of these results requires further verification in more diverse populations. Furthermore, this study did not cover unknown or uncultivated microbial flora, which may limit our understanding of the full complexity of microbial communities. Future research should aim to cultivate and include these unclassified microbial groups to further explore their mechanisms of action, with the expectation of providing more valuable information. Lastly, although we have made causal inferences, it is important to acknowledge that MR methods, while providing strong evidence for causality, still rely on statistical assumptions that may not hold in all cases. Therefore, while our results suggest a potential causal link, further experimental validation and additional studies are needed to confirm these findings. Despite these limitations, this study, as the first MR research exploring the causal relationship between skin microbial communities and T2DM, remains pioneering. Our findings suggest that skin microbiota may serve as potential biomarkers for T2DM, offering a basis for personalized prevention strategies targeting the microbiome, such as through diet or probiotics. Additionally, by identifying the role of microbial-driven inflammation in T2DM, we propose that future treatments could focus on modulating the skin microbiota to reduce inflammation and improve insulin sensitivity. These insights pave the way for microbiota-based approaches in early detection, personalized medicine, and targeted therapies for T2DM.






Comments (0)