
Remember me
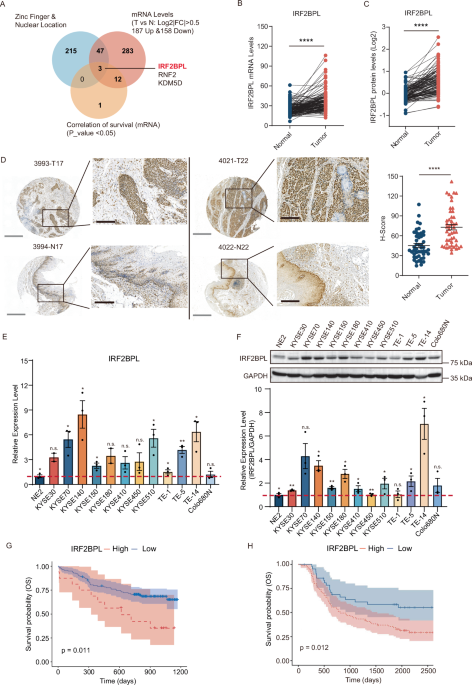
To check whether expression of Erk1R84H may give rise to tumors in vivo, we established a transgenic mouse model that allows tissue-specific and temporally controlled expression of Erk1R84H. While the model allows expression in any tissue, this study is focused on the liver. The RTK-Ras-Raf-MEK-Erk pathway is abnormally active in 30% of liver cancer cases and in ~80% of liver cancer in mice induced by diethylnitrosamine (DEN) [4, 48], suggesting that the liver is a suitable tissue for testing whether activation of Erk alone could cause cancer. The design of the transgenic system that allows liver-specific expression when doxycycline (dox) is provided in the diet, is described in detail in Supplementary File S1 and in Fig. 1A. Mice harboring one allele of the expression cassette were termed hepatic-hetero-Erk1R84H, while those harboring two alleles were termed hepatic-homo-Erk1R84H mice. The levels of Erk, pErk and pT207 were compared between the hepatic-hetero-Erk1R84H and the hepatic-homo-Erk1R84H mice (Supplementary Fig. S1).
Fig. 1: Design, establishment and confirmation of the transgenic mouse model that inducibly expresses Erk1R84H specifically in the liver.
A Schematic representation of the transgenic mouse model. The upper part of the panel shows the two expression cassettes inserted at the Rosa26 locus of the carrier mice. In the cassette on the left the rtTA coding sequence (rtTA-M2) is transcriptionally controlled by the strong and constitutively active CAGGS promoter, but a transcription intervening sequence (STOP), that is bordered by flox sequences, separates between the promoter and the gene. Therefore, the gene encoding the rtTA-M2 could be transcribed only if the “STOP” is removed. In the cassette on the right the cDNA encoding flag-tagged Erk1R84H could be transcribed under the tetO Pcmv-promoter, which is activated by rtTA-M2. The second row of the panel shows the driver mouse in which Cre recombinase is expressed only in the liver, under the albumin promoter. In progeny of crossbreeding between the carrier mice and the Alb-Cre driver mice (middle panel), termed hepatic-hetero-Erk1R84H mice, the liver-specific expression of Cre recombinase causes excision of the STOP sequence in hepatocytes only and consequently transcription of rtTA-M2. Upon doxycycline (dox) administration (bottom panel), dox-bound rtTA-M2 is active as a transcriptional activator and activates the tetO Pcmv-promoter that drives expression of 3xFlag-tagged Erk1R84H specifically in the liver. B Western blots analysis, with the indicated antibodies, of protein lysates prepared from various organs of hepatic-hetero-Erk1R84H mice fed with dox-supplemented diet for 2 weeks.
To check the operability of the model, hepatic-hetero-Erk1R84H mice were fed with a dox-supplemented diet for 2 weeks, and various organs were collected to check for Erk1R84H expression. Expression of Erk1R84H was observed only in the liver, and only in mice provided with dox-supplemented diet, validating the model design (Fig. 1B). Importantly, the transgenic Erk1R84H protein was phosphorylated on the TEY motif, demonstrating that in the context of the whole organism it maintained the property of becoming spontaneously active. Notably, the phosphorylation level was not very high (Fig. 1B, row 2), as a consequence of powerful feedback inhibition as will be explained in detail below. As expected from in vitro and tissue cultures studies [44, 46], Erk1R84H was also autophosphorylated on Thr207 in the liver (Fig. 1B, row 3), a phosphorylation reported to be associated with human diseases [49]. To our knowledge, this is the first model that allows activation of Erk molecules per se (i.e, without activating upstream components) in vivo, in tissue-specific and temporally controlled manners.
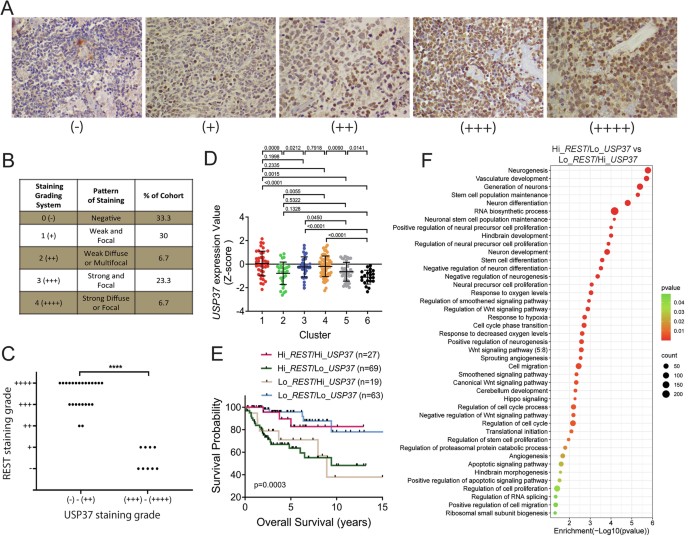
Liver-specific expression of Erk1R84H causes development of hepatocellular carcinoma (HCC)To assess whether expression of Erk1R84H affects the liver in any way, groups of hepatic-hetero-Erk1R84H and hepatic-homo-Erk1R84H mice were provided with a dox-supplemented diet (to induce liver-specific Erk1R84H expression) while other groups of the same mice were provided with a regular diet. Mice were randomly selected for each group. It was noted that about 12 weeks after the beginning of the experiment both hepatic-hetero-Erk1R84H and hepatic-homo-Erk1R84H mice provided with dox-supplemented diet began to lose weight and continued to lose weight until the end of the experiment (Fig. 2A). During the course of the experiment mice were sacrificed at 111, 152, and 182 days (3.7, 5 and 6 months respectively) after change of diet, and the livers were removed for analysis.
Fig. 2: Liver-specific Erk1R84H expression causes hepatocellular carcinoma (HCC).
A Changes in total body weight of hepatic-Erk1R84H mice fed with a regular diet (black circles; n = 31) or dox-supplemented diet (red circles; n = 30). Data are shown as mean ± SEM. B Macroscopic appearance of livers from hepatic-homo-Erk1R84H mice provided with dox-supplemented diet for 5 months (two photos on the left) showing signs of significant deformations, and of a hepatic-homo-Erk1R84H mouse provided with regular diet for the same time (photo on the right). C As in B but after tissue fixation. D Liver-to-body weight ratio in hepatic-homo-Erk1R84H mice provided with a regular diet (black circles) or a dox-supplemented diet (for inducing liver-specific expression of Erk1R84H) (red circles). Data are represented as mean ± SEM. Statistical significance was determined by unpaired two-tailed t test with significance indicated by ****, representing a P-value of p < 0.0001. E Total liver weight of hepatic-homo-Erk1R84H mice fed dox-supplemented diet (red circles) or regular diet (black circles). Data are represented as mean ± SEM. Statistical significance was determined by unpaired two-tailed t test with significance indicated by ****, representing a P-value of p < 0.0001. Notably, investigators were not blinded to the experimental groups.
Inspection of the livers of hepatic-homo-Erk1R84H mice provided with dox-supplemented diet for 5 months revealed dramatic deformation, including areas of discoloration and nodular growths, which are suggestive of tumor-like bodies (Fig. 2B, C). There was a significant portion of the liver that looked darker, possibly indicating areas of necrosis or hemorrhage. The surface of the liver was not uniform, but rather had multiple swollen areas which could be indicative of lesions or tumors. These irregularities were in stark contrast to the smooth, homogenous and overall normal appearance of the livers removed at the same timepoints from similar mice not expressing the active Erk1 (Fig. 2B, C). Also, at all timepoints the liver to body weight ratio of hepatic-homo-Erk1R84H was markedly higher compared to that in mice not expressing it (Fig. 2D). The mean of liver weight of mice not expressing Erk1R84H was 1.68+/− 0.24 mg while the liver weight mean of mice expressing Erk1R84H was 2.61 +/− 0.68 mg (Fig. 2E).
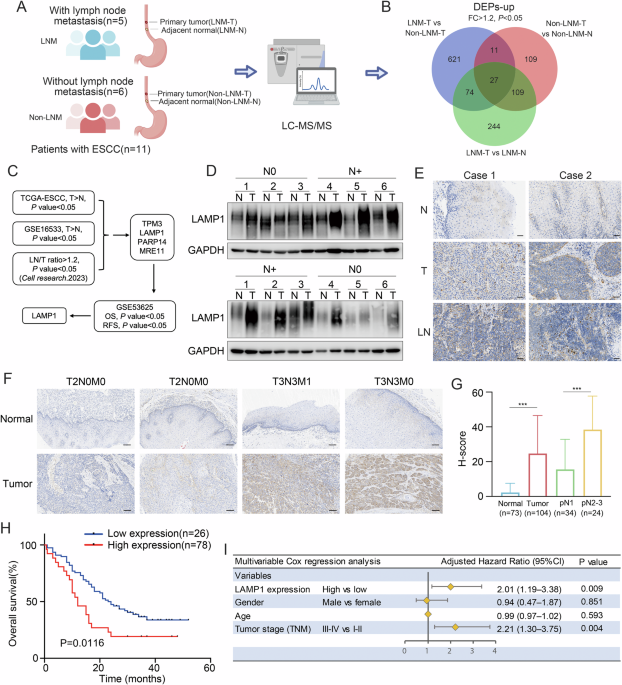
Pathological examination of hematoxylin and eosin (H&E)-stained cross sections revealed that liver was seriously affected in 100% of both heterozygous or homozygous, upon induction of Erk1R84H. Notable alterations in livers of hepatic-homo-Erk1R84H mice expressing the transgene for 111 days (3.7 months) included increased basophilia, pronounced anisokaryosis, and varying cytoplasmic volumes, as illustrated in Fig. 3A1. None of these phenotypes were observed in livers of identical hepatic-homo-Erk1R84H mice not expressing Erk1R84H (Fig. 3A3). Additionally, livers expressing Erk1R84H exhibited heterogeneity in cellular density and occasional presence of deeply eosinophilic cytoplasmic inclusions, as well as areas of coagulation necrosis (Fig. 3A2). These phenotypes suggest potential liver dysfunction.
Fig. 3: Pathological analysis of livers from hepatic-Erk1R84H transgenic mice reveals tumor development and hepatic abnormalities.
A Representative H&E staining of liver cross sections from hepatic-homo Erk1R84H mice provided with dox-supplemented diet for 3.7 months (1–2) or 5 months (4–9), or with regular diet for the same periods (3 and 10) (scale bar = 200 µM). B An enlarged view of a liver section from a hepatic-homo mouse expressing Erk1R84H for 5 months, highlighting increased mitotic activity (reflected by chromosomes at various stages of mitosis; circled regions) as a marker of uncontrolled cell proliferation, as is common in HCC (scale bar = 200 µM).
Pathological analysis of livers removed from hepatic-homo-Erk1R84H mice expressing Erk1R84H for more than 5 months revealed an increased tendency for tumor development. Specifically, 54.5% of the livers (6 out of 11) exhibited lesions ranging from moderately to poorly differentiated hepatocellular carcinoma (HCC) (Fig. 3A4-9). Liver sections prepared from these mice showed an increased mitosis rate (Fig. 3B), irregular foci of proliferation (Fig. 3A4, A5), and areas of hemorrhage and coagulation necrosis (Fig. 3A6, A7). These phenotypes were not observed in liver sections from hepatic-homo-Erk1R84H mice fed a regular diet (i. e. not expressing Erk1R84H) for the same period (Fig. 3A10). The 5 mice that did not manifest clear HCC phenotypes displayed anisokaryosis (Fig. 3A8) and accumulation of eosinophilic cytoplasmic globules (Fig. 3A9), similar to the phenotype manifested by the mice expressing Erk1R84H for 3.7 months.
The phenotype of hepatic-hetero-Erk1R84H mice that were on a dox-supplemented diet for about 5 months was less severe than that of the hepatic-homo-Erk1R84H mice with respect to tumor development. Nevertheless, they did demonstrate an inclination towards dilation of arteries, suggesting a problem with blood pressure (Supplementary Fig. S2), and exhibited similar phenotypes to those seen in hepatic-homo-Erk1R84H mice with respect to accumulation of eosinophilic globules and anisokaryosis (Supplementary Fig. S2A, B). It appears that hepatic-hetero-Erk1R84H mice fed on a dox-supplemented diet represent a stage where the tissue is inclined or prone to become cancerous, while many of the hepatic-homo-Erk1R84H mice, having two alleles of Erk1R84H develop the full-scale disease. Importantly, liver disease was significant and prominent in 100% of the mice expressing the active Erk1R84H, whether heterozygous or homozygous, compared to 0% among those fed with a regular diet and therefore did not express the active Erk1R84H. The difference between heterozygous and homozygous expression is expected, as noted in other mouse models that used the same double-cassette expression system [50, 51] (see discussion).
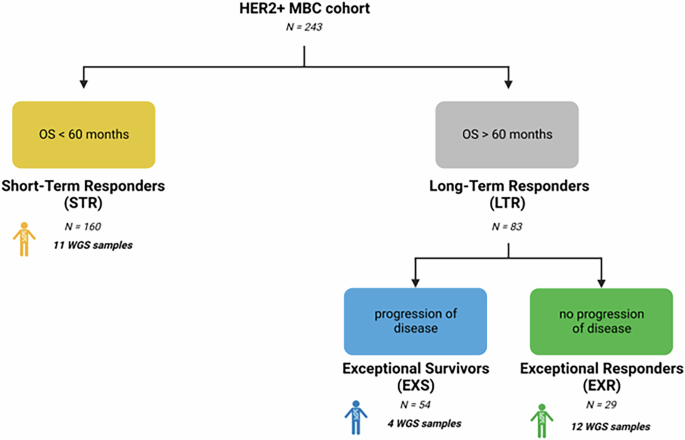
Kidneys are also significantly affected in mice expressing Erk1R84H in the liverWhile removing the livers of mice expressing Erk1R84H we noted the abnormal appearance of the kidney. Kidneys were smaller in size and somewhat paler compared to those from mice not expressing the active Erk1R84H variant (Fig. 4A). We thus examined kidneys of homozygous mice expressing Erk1R84H for 3 months. The pathologist’s report highlighted several renal pathologies, including the presence of protein casts, glomerular thickening, and perivascular infiltration (Fig. 4B), phenotypes that were not observed in a similar mouse not expressing Erk1R84H in the liver (Fig. 4C). These findings suggest a serious renal disorder causing both functional and structural kidney alterations.
Fig. 4: Kidney are altered in hepatic-Erk1R84H mice although Erk1R84H is expressed exclusively in the liver.
A Macroscopic morphology of kidneys of hepatic-homo-Erk1R84H mouse expressing (right) or not expressing (left) Erk1R84H for 3 months. B Representative H&E-stained section of kidneys from hepatic-homo Erk1R84H mice supplemented with dox-supplemented diet for 3 months showing kidney abnormalities. C As in (B) but sections from a control mouse, fed with regular diet, showing a normal kidney appearance. D A western blot analysis of protein lysates from various organs of hepatic-homo-Erk1R84H mouse expressing Erk1R84H for 5 months. Note that Erk1R84H expression was restricted to the liver. No Erk1R84H expression was detected in the kidney.
The renal changes are probably a secondary effect of hepatic dysfunction caused by the specific expression of Erk1R84H in the liver (Supplementary Fig. S3). This conclusion is based on the analysis of serum markers indicative of both hepatic and renal function, which revealed that liver-associated markers (e.g., albumin, bilirubin, total protein, calcium) were altered at both early and late time points, whereas kidney-associated markers (e.g., urea and creatinine) showed changes only at later time points (Supplementary Fig. S3). The protein casts in the kidneys, which represent protein accumulation, might originate from protein accumulation in the liver, implying a deficiency in hepatic protein processing and subsequent accumulation in the kidneys. Importantly, despite the dramatic renal effects observed, we confirmed that Erk1R84H expression was confined to the liver, with no detectable expression in the kidneys – even in mice expressing Erk1R84H for 5 months, as indicated by α-Erk antibody (Fig. 4D). The temporal pattern of serum marker changes, combined with the absence of Erk1R84H expression in the kidneys, supports the hypothesis that the renal pathology arises as a secondary consequence of sustained liver dysfunction.
TEY phosphorylation of Erk1R84H is shut off during the development of liver cancerTo check for the linkage between the activity of Erk1R84H and oncogenic transformation of the liver, we followed the status of Erk1R84H phosphorylation for 190 days. Similar to the case in the experiment shown in Fig. 1B, in this experiment too, TEY was highly phosphorylated in mice expressing Erk1R84H for 14 days. However, the phosphorylation levels were significantly lower as early as 33 days after induction of expression and remained at the edge of detectability throughout the experiment (Fig. 5A). Namely, as liver pathology progressed, Erk1R84H’s phosphorylation became extremely low, and remained barely detectable in the cancerous liver (Fig. 5A). Notably, phosphorylation of endogenous Erk1 and Erk2 molecules was also downregulated (Fig. 5A, row 2). Similar kinetics of pErk1R84H downregulation were observed in the hepatic-homo-Erk1R84H model (Supplementary Fig. S4), further supporting the generality of this phenomenon. These findings suggest that tumors are maintained and continue to grow despite the markedly low catalytic activity of the transforming oncogene.
Fig. 5: Dramatic downregulation of TEY phosphorylation of Erk1R84H and endogenous Erk1/2 during the development of liver cancer.
A Western blot analysis of liver protein lysates from hepatic-Erk1R84H mice provided with dox-supplemented diet for the indicated time periods. B Western blot analysis of livers of hepatic-Erk1R84H mice grown on regular diet (1st and 4th lanes from the left), or dox-supplemented diet (2nd lane from left) for six months, or on dox-supplemented diet for 5 months and then on regular diet for 1 month (3rd and 5th lanes from the left).
We next asked whether the downregulation in TEY phosphorylation in the hepatic-Erk1R84H mouse model is a consequence of a new biochemical activity that emerged in the transformed hepatocyte or is still dependent on the activity of Erk1R84H. In other words, would the vanished TEY phosphorylation re-appear if Erk1R84H expression would be shut-off? To explore this question, we used hepatic-hetero-Erk1R84H mice expressing Erk1R84H for 5 months, replaced the dox-supplemented diet with a regular diet and sacrificed the mice one month later. As is shown in Fig. 5B, while expression of Erk1R84H was downregulated upon transfer to a regular diet, TEY phosphorylation of both Erk1R84H and endogenous Erk1/2 was concomitantly elevated (Fig. 5B, columns 3 & 5, row 2). This observation underscores the reliability of our transgenic mouse model and strongly suggests that the downregulation of TEY phosphorylation of Erk1R84H is a direct consequence of its own activity.
In the process of transforming NIH3T3 cells, phosphorylation of the oncoprotein Erk1R84H is dramatically reduced and becomes undetectable in the transformed cellsTo explore the oncogenic mechanism utilized by Erk1R84H, to test whether it is sensitive to Erk inhibitors and to explore the shut-off mechanism, we used NIH3T3 cells, which can be transformed by this molecule [44]. We first followed the status of Erk1R84H in cells transfected with a plasmid expressing 3Xflagged-Erk1R84H. A plasmid expressing 3Xflagged-Erk1WT was transfected in parallel as a control. G418 selection was imposed 24 h post transfection and samples of cells were harvested at 2, 5, 10, 15 and 20 days post transfection. Western blot analysis showed that Erk1R84H was catalytically active, as manifested by its strong phosphorylation on the TEY motif at day 2 post transfection. It remained phosphorylated, though to lower levels, for 5 days after transfection (Fig. 6A, row 2). Phosphorylation level of Erk1WT was low at these timepoints (Fig. 6B, second row). As expected, due to the G418 selection process and death of cells not harboring the plasmids, from day 5 onward Erk1R84H expression levels decreased, and accordingly, in day 10 phosphorylation was almost undetectable (Fig. 6A, rows 1 & 2). While expression of Erk1R84H was clearly observed at days 15 and 20 post-transfection (as at this timepoint most cells are G418-resistant, i.e., harboring the expressing plasmid) its phosphorylation was almost undetectable (Fig. 6A). Phosphorylation of Thr207 was also significantly downregulated from day 2 to day 20 (Fig. 6A, row 3). These intriguing results are reminiscent of the observation in the hepatic-Erk1R84H mouse model, as between day 15 and 20 post transfection, when Erk1R84H phosphorylation is barely detectable, transformed foci develop in the plate.
Fig. 6: Oncogenic transformation of NIH3T3 cells by Erk1R84H is also accompanied by downregulation of TEY phosphorylation.
Western blot analysis of lysates prepared from NIH3T3 cells transfected with a plasmid expressing 3Xflagged-Erk1R84H (A) or with 3Xflagged-Erk1WT (B) at the indicated timepoints following transfection.
Unlike the case of Erk1R84H, expression of Erk1WT, which was reduced at day 10, did not recover, and remained low at day 20 (Fig. 6B, first row). As explained, Thr-207 phosphorylation is exclusive to active variants and therefore not detected in cells transfected with a vector that drives Erk1WT expression.
As in the hepatic-Erk1R84H mouse model Erks’ phosphorylation was almost undetectable in the tumors themselves, we also examined its phosphorylation in stable clones expressing Erk1WT or Erk1R84H. In this experiment we also included cells transformed with Erk1R84S (note that clones expressing Erk1WT are derived from G418-resistant colonies while clones expressing Erk1R84S or Erk1R84H are derived from foci). Notably, clones expressing Erk1R84H or Erk1R84S remained transformed following 5 passages or more, as well as following freezing and thawing steps, continuing to exhibit altered cell morphology and enhanced growth rate. As reported previously, a clear evidence for the maintenance of the transformed phenotype is the ability of these cells, after several passages, and taken from frozen stocks, to give rise to tumors in nude mice [46]. Despite their transformed state, levels of TEY phosphorylation of Erk1R84H and Erk1R84S were consistently below detection levels in these clones, although the molecules were clearly expressed (Fig. 7A, three left lanes). Thus, similar to the case in mice, cells transformed by Erk1R84H or Erk1R84S maintain their oncogenically-transformed phenotype despite the markedly low catalytic activity of the transforming oncogene. Intriguingly, not only Erk1R84H and Erk1R84S were not phosphorylated in cells transformed by them, but also phosphorylation of endogenous Erk1/2 was very low in these cells (Fig. 7A, row 2). Thus, in both hepatic-Erk1R84H mice and in NIH3T3 cells, the machinery that suppresses Erk1/2 phosphorylation upon Erk1R84H expression is very efficient, affecting auto-phosphorylated Erk molecules (the active variants), and also those phosphorylated by the RTK-Ras-Raf-MEK cascade (endogenous Erk1/2). In stable clones harboring the vector expressing Erk1WT, the observation is puzzling. The exogenous Erk1WT is expressed at a relative low level and is not active (Fig. 7A, lanes 1). However, the phosphorylation of the endogenous Erks is still suppressed, as in cells expressing Erk1R84H and Erk1R84S. Perhaps some expression levels of Erk1WT, higher than in native levels, are sufficient to activate the machinery that suppresses Erk phosphorylation.
Fig. 7: Suppression of Erk1R84H phosphorylation occurs via a powerful feedback mechanism as it is relieved by Erk inhibitors.
A Western blot analysis of protein lysates prepared from stable NIH3T3 clones expressing Erk1WT, Erk1R84S, or Erk1R84H 3 days post plating of frozen cells, without (left three lanes) or with (right three lanes) exposure to 5 μM BVD-523 for 2 h. B As in A but lysates were prepared from cells 24 h post plating of frozen cells. Western blot analysis of lysates prepared from NIH3T3 cells transfected with a plasmid expressing Erk1R84H (C) or Erk1WT (D) at the indicated timepoints post transfection (5 lanes from the left), and of cultures provided with 5 μM BVD-523 or DMSO for 2 h at day 20 post transfection (the two lanes at the right).
We further monitored TEY phosphorylation at different stages of the life of the stable clones and found that at particular short time-windows in the culture life, when cells are plated sparsely, or when cells are just thawed from a frozen stock (no longer than 24 h after thawing) TEY phosphorylation could be detected. Note three left lanes in Fig. 7B, which shows analysis of a culture 24 h after thawing (compare to three left lanes in 7 A). At all other time Erk TEY phosphorylation is strongly suppressed.
Suppression of Erk1R84H phosphorylation occurs via a powerful feedback mechanism that is part of the oncogenic processWe next wished to test whether in culture too, phosphorylation of Erk could be restored by suppression of the overactive Erk1R84H, and thereby confirming that the potent downregulation of the active form of Erk1R84H is directly linked to its own activity. A possible way of reducing Erk1R84H activity could be through the use of Erk’s pharmacological inhibitors. The rationale is that if it is the high activity of Erk1R84H or Erk1R84S that negatively regulates their own phosphorylation then its inhibition would restore phosphorylation.
This experimental approach is valid, however, only if Erk1R84H and Erk1R84S are proven to be sensitive to Erk inhibitors, which were developed against MEK-activated Erks and not against autoactivating molecules [22, 29, 52, 53]. We thus first performed a series of systematic experiments, in which we found that the oncogenic molecules Erk1R84H and Erk1R84S maintain sensitivity to several Erk-specific inhibitors, including BVD-523 (described in Supplementary File S2).
Having verified that Erk1R84H and Erk1R84S are sensitive to the pharmacological inhibitors (Supplementary File S2), we exposed foci-derived clones, stably expressing Erk1R84H or Erk1R84S, to BVD-523. This treatment caused strong phosphorylation of the TEY motif of all Erk molecules in the cells (Fig. 7A and 7B, 3 right lanes). The response of Erk molecules was so rapid that within 2 h of treatment the signal was detected (Fig. 7A). While exposure to BVD-523 boosted TEY-phosphorylation, it decreased Thr207 phosphorylation of Erk1R84H (Fig. 7A, B, 3rd row). Given that Thr207 phosphorylation is a consequence of autophosphorylation [44, 46], its downregulation is expected once the catalytic activity of Erk1R84S and Erk1R84H is inhibited.
Exposure to BVD-523 also elevated TEY phosphorylation in stable clones harboring plasmid carrying Erk1WT (Fig. 7A, B, 4th column), suggesting that this molecule also imposes some degree of negative feedback inhibition.
To test whether the strong induction of Erk’s phosphorylation is a general phenomenon upon Erks’ inhibition, and not exclusive to BVD-523, the stable clones were exposed to SCH772984 and GDC0994. Similar to BVD-523, GDC0994 restored Erk’s phosphorylation (Supplementary Fig. S5, columns 4 and 9). SCH772984 did not affect Erk phosphorylation, probably due to its different mechanism of inhibition that interferes with Erk phosphorylation by MEK (see Discussion) (Supplementary Fig. S5, columns 3 and 8).
We next wondered whether the downregulation of Erk observed during the very early oncogenic transformation stages is also mediated via feedback inhibition, as was the case in the stable clones. For that we repeated the transfection experiment, and at day 20, when the shut-off of Erk phosphorylation is apparent, exposed cells to BVD-523 for 2 h. This treatment caused significant elevation of TEY phosphorylation of the Erk1R84H protein, as well as of endogenous Erk1 and Erk2 molecules (Fig. 7C). Thr-207 phosphorylation was not affected (Fig. 7C, row 3). The effect of BVD-523 suggests that the rapid disappearance of TEY phosphorylation in cells expressing Erk1R84H and Erk1R84S is a consequence of the unregulated activity of these molecules, which most probably inhibit pathway’s upstream components and also activates negative regulators such as phosphatases (confirmed by proteomic analysis, see below; see Fig. 11D). Once Erk1R84H and Erk1WT are inhibited by the pharmacological reagents, this feedback inhibition is relieved, leading to elevated phosphorylation of MEKs (Fig. 7C, row 5 and Fig. 7D, row 4) and in turn to MEK-mediated Erk phosphorylation (Fig. 7C, D, row 2). The fact that the feedback inhibition is so profound as early as 20 days after transfection, suggests that Erk1R84H and Erk1R84S inhibition is an early step in the oncogenic sequela.
Although active Erks are shut-off in the course of oncogenic transformation, Erk inhibitors prevent transformationThe observation that Erk1R84S and Erk1R84H seem to be almost hermetically shut-off in the course of transformation and remain suppressed when the transformed phenotype is maintained, raises the question of whether their catalytic activity is at all required for the oncogenic process. To address this riddle, we tested the ability of Erk1R84S and Erk1R84H to transform NIH3T3 cells in the presence of inhibitors. NIH3T3 cells were transfected with plasmids expressing Erk1WT, Erk1R84H, Erk1R84S, or with an “empty” plasmid. 24 h after transfection cells were provided with G418 and with 0.1 or 0.5 μM SCH772984, GDC-0994, or BVD-523. Media containing the inhibitors was changed every other day and the cells were monitored for a period of 30 days. In plates not exposed to inhibitors and expressing Erk1R84H or Erk1R84Sfoci appeared as expected (Fig. 8). In plates supplemented with inhibitors, foci were commonly not observed, suggesting that inhibiting the catalytic activity of Erk1R84H or Erk1R84S prevented the oncogenic process (Fig. 8). This implies that Erk1R84S and Erk1R84H activity is critical for transformation, even though it is shut-off at early stages of the process. It should be noted that at the concentrations used the inhibitors were not toxic to the cultures and cells of all clones did proliferate and fill the plates. The effect was specific to the formation of foci (Fig. 8).
Fig. 8: Erks’ pharmacological inhibitors prevent oncogenic transformation of NIH3T3 cells by Erk1R84S and Erk1R84H.
Crystal violet staining of NIH3T3 cells transfected with plasmids expressing Erk1WT, Erk1R84S, Erk1R84H, or an “empty” plasmid. 24 h post-transfection, cells were subjected to G418 selection. Erk inhibitors were introduced 48 h post-transfection. Staining was performed 30 days post-transfection to assess cell viability and focus formation.
Notably, this experiment disclosed a difference in the response to inhibitors between Erk1R84S and Erk1R84H. While 0.1 μM of SCH772984 or BVD-523 was sufficient to completely inhibit the appearance of foci in Erk1R84S-transfected cells, it was not sufficient to absolutely eliminate foci development by Erk1R84H (Fig. 8). It seems that Erk1R84H is a more robust and aggressive onco-protein than Erk1R84S.
Next, we examined the effect of introducing inhibitors at later stage post-transfection to assess whether relieving the negative feedback could halt or regress tumor progression. 5 μM of BVD-523 was thus added 13 days post-transfection, at a time when small foci had already formed. As shown in Fig. 9, the introduction of inhibitors at this later stage effectively blocked further progression of the foci (Fig. 9).
Fig. 9: Introduction of Erk inhibitors at a late stage post-transfection stops further development of transformed foci.
NIH3T3 cells transfected with Erk1R84H were allowed to grow for 13 days until small foci were visible, at which point 5 μM of BVD-523 was added to the media. Cultures were stained with crystal violet 20 days post-transfection.
Erk proteins are also shut-off via a negative feedback machinery in cells derived from human cancerAs described in the introduction, various levels of phosphorylated Erks were reported in different cancers [37,38,39,40,41]. These very different levels were proposed to reflect the resultant interplay between activation and feedback inhibition pathways of the RTK-Ras-Raf-MEK-Erk cascade [54,55,56,57,58,
Comments (0)