
Remember me

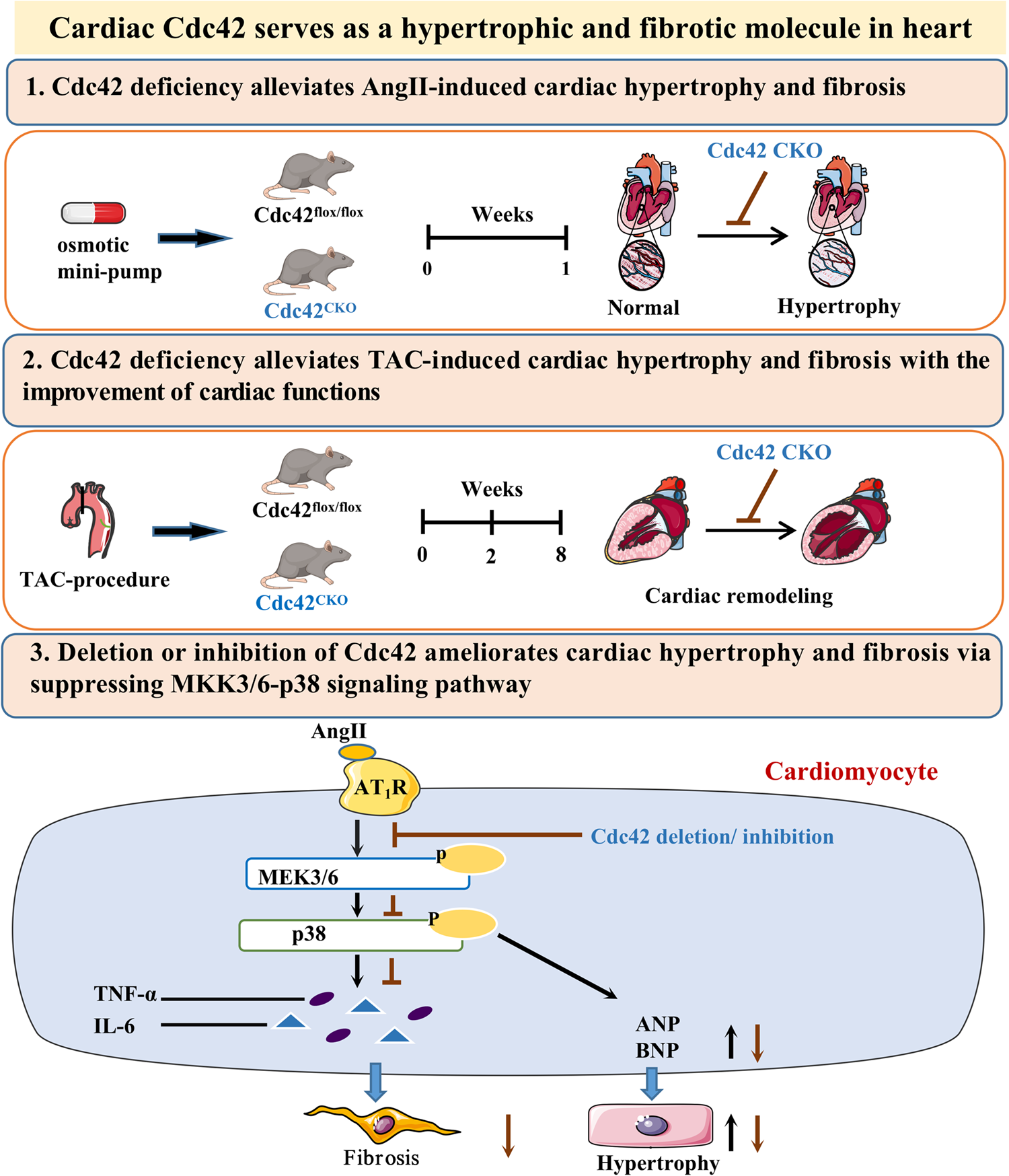
We previously reported that the human MyD88 adaptor forms cytosolic spots that localize to ER-mitochondria membrane junctions when expressed in S. cerevisiae [24]. Given the controversy surrounding the subcellular localization of MyD88 in mammalian cells prior to TLR stimulation [18, 34], we aimed to further investigate this aspect in yeast. To this end, we generated a series of MyD88 truncations fused to fluorescent proteins to assess the contribution of its individual domains to this localization pattern.
Firstly, we designed truncated MyD88 variants that retained the Death domain (DD) in all cases, as this domain is essential for myddosome oligomerization [19]. Three variants were generated (Fig. 1a): one lacking the TIR domain and spanning amino acids 1-160 (N-DD-INT), another additionally lacking the INT domain and comprising amino acids 1-109 (N-DD), and the MyD88-S splicing variant, which lacks the INT domain [8]. Overproduction of these MyD88 versions, like that of the WT protein, did not affect yeast growth, as determined by a drop growth assay (Fig. S1a). As shown in Fig. 1b, immunoblot analysis revealed variations in the expression levels of the different truncated MyD88-EGFP versions. The N-DD and MyD88-S variants exhibited lower protein levels compared to the N-DD-INT and WT versions, with a particularly pronounced reduction in N-DD. This suggests that the TIR and, especially, the INT domain contribute to protein stability.
Fig. 1
The absence of the INT and/or TIR domains reduces MyD88 stability in yeast while preserving ERMES localization. (a) Schematic representation of the different MyD88 variants: the wild type protein (MyD88), a version lacking the TIR domain (N-DD-INT), one lacking both the TIR and INT domains (N-DD), and the splicing variant naturally occurring in human cells, lacking only the INT domain (MyD88-S). Domain color coding: orange for the N-terminal region, purple for the Death domain, blue for the INT domain and red for the TIR domain. (b) Immunoblot on cell extracts from the YPH499 strain transformed with plasmids pAG425-Ø-EGFP, pAG425-MyD88-EGFP, pAG425-MyD88-N-DD-INT-EGFP, pAG425-MyD88-N-DD-EGFP, or pAG425-MyD88-S-EGFP. Heterologous proteins were detected using an anti-GFP antibody, and the G6PDH protein was used as a loading control. The quantitative data shown represent the ratio of each band’s intensity relative to the loading control, normalized to wild-type MyD88. (c) Differential Interference Contrast (DIC) and fluorescence microscopy images of YPH499 cells co-transformed with plasmids pAG424-Mdm34-DsRed and pAG425-MyD88-EGFP, pAG425-MyD88-N-DD-INT-EGFP, pAG425-MyD88-N-DD-EGFP, or pAG425-MyD88-S-EGFP. The experiment was performed as a biological triplicate, and representative images are shown. Scale bar: 5 μm. (d) Bar graph showing the percentage of cells displaying detectable green fluorescence corresponding to MyD88 or its variants. Excitation conditions were identical for all four samples, and brightness and contrast were varied equally in all images. The experiment was performed as a biological triplicate and > 50 cells of each transformant were analyzed. Error bars represent the SD. Asterisks indicate a p value < 0.001 (***) and < 0.05 (*), calculated using Tukey’s HSD test
The elimination of the TIR and/or INT domains of MyD88 did not modify the subcellular localization pattern, which remained identical to that of the full-length protein, colocalizing with the ERMES component Mdm34 tagged with DsRed (Fig. 1c). However, consistently with the observed loss of protein stability, a significant reduction in the number of cells displaying MyD88 spots was detected (Fig. 1d).
Deletion of residues 1–20 upstream MyD88 Death domain shifts its aggregation from spots to filamentsThe complete three-dimensional structure of MyD88 has not yet been solved experimentally. To gain further insight into its structure, we performed structural modeling of the full-length protein using ColabFold v1.5.3-patch [31]. In the prediction model shown in Fig. 2a, the Death and TIR domains are resolved with a high level of confidence (plDDT > 90, or < 90 and > 70). However, the N-terminal region, corresponding to the first 20 amino acids, as well as certain fragments of the INT domain, exhibit significantly lower confidence scores (plDDT < 70 and > 50, or < 50). This may reflect that these are disordered regions, less conserved in evolution as the Death and TIR domains. Indeed, a multiple sequence alignment of the MyD88 sequence from 25 metazoan species reveals that a reliable consensus sequence can only be established starting at position 20 in the human MyD88 sequence. Furthermore, we observed some correlation between the extension of the N-terminal region and the evolutionary branch within vertebrates: the extension is generally longer in mammals, intermediate in birds, reptiles, and amphibians, and shorter in fish (Fig. S2).
Fig. 2
The first 20 amino acids at the N-terminus of human MyD88 influence its stability and homopolymerization pattern. (a) Predicted three-dimensional structure of MyD88 generated using ColabFold v1.5.3-patch. Amino acid residues are colored according to the confidence of the prediction, based on the plDDT (predicted local distance difference test) score. The model has an average plDDT of 83.4 and a pTM-score of 0.521. A total of 6 recycling steps were applied during the prediction. (b) Immunoblot of cell extracts from YPH499 strain transformed with plasmids pAG425-Ø-EGFP, pAG425-MyD88-EGFP, pAG425-MyD88-N1-20-EGFP, pAG425-MyD88-∆N1-20-EGFP or pAG425-MyD88-∆N1-7-EGFP. Heterologous proteins were detected using an anti-GFP antibody, with G6PDH as the loading control. Quantification shows the ratio of each band intensity relative to the loading control, normalized to wild-type MyD88-EGFP. (c) DIC and fluorescence microscopy images of YPH499 cells transformed with the same plasmids as in panel (b). Representative fields are shown. A graph is shown displaying the percentage of cells with distinct MyD88 subcellular localizations, as noted. Error bars represent the SD from three different biological replicates (n≈100 for each transformant clone). Statistical significance of cells with a single spot on WT MyD88 vs. all truncations (blue), as well as cells with filamentous structures in MyD88-∆N1-20 vs. the rest (green) are indicated with a p-value < 0.001 (***), according to Student t-test analyses. (d) Fluorescence microscopy images of YPH499 cells co-transformed with plasmids pGPD416-mCherry-Lact-C2 and pAG425-MyD88-∆N1-20-EGFP (top panels); pSM1959-Sect. 63-mRFP and pAG426-MyD88-ΔN1-20-EGFP (middle); or YEplac112-Ilv6-mCherry and pAG425-MyD88-ΔN1-20-EGFP (bottom). (e) Fluorescence microscopy images of YPH499 cells co-transformed with pAG424-Mdm34-DsRed and pAG425-MyD88-ΔN1-20-EGFP. All microscopy experiments were performed with biological triplicates, and representative images are shown. Scale bars represent 5 μm. (f) Predicted three-dimensional structures of four MyD88 molecules (left panel) and four MyD88-∆N(1–20) molecules (right panel) generated with ColabFold v1.5.5. Each monomer is shown in a different color (red, green, blue, purple). The MyD88 × 4 model has an average plDDT of 66.6, a pTM-score of 0.333, and an ipTM-score of 0.254. On the other hand, the ∆N1-20 × 4 model has an average plDDT of 71.8, with the pTM-score and ipTM-score values of 0.437 and 0.376, respectively. Three recycling steps were applied for both models
Considering this information, along with previous studies suggesting that the N-terminal region of MyD88 is responsible for its subcellular localization [18, 35], we expressed two truncated versions of MyD88 in yeast: one lacking the first 7 amino acids (MyD88-∆N1-7), corresponding to the shortest MyD88 sequences found in fish, and another lacking the whole 20 amino acid N-terminal extension (MyD88-∆N1-20). Additionally, the first 20 amino acids of MyD88 were cloned separately (N1-20) to assess whether this segment alone was sufficient to drive a specific subcellular localization pattern in yeast. Like WT MyD88, expression of these truncated variants did not cause toxicity in yeast (Fig. S1b). Remarkably, as shown in Fig. 2b, the MyD88-ΔN1-20 EGFP fusion was immunodetected from yeast lysates to a greater extent than that of full-length MyD88, whereas MyD88-∆N1-7 yielded a fainter band. Regarding subcellular localization, the ∆N1-20 variant predominantly formed cytoplasmic filaments instead of compact puncta in a significant proportion of cells (51.71 ± 7.06%), a behavior never observed for full-length MyD88, as well as cells with more than one fluorescent spot (42.77 ± 5.31) (Fig. 2c). In contrast, the ∆N1-7 variant significantly lost the characteristic punctate pattern (spots were only observed 8.46 ± 1.50% of the cells). The construction comprising only the first 20 amino acids of MyD88, N(1–20), presented a diffuse cytosolic pattern in 100% of the cells (Fig. 2c). These results indicate that such N-terminal extension itself is not responsible for the subcellular localization of MyD88 in yeast cells. Moreover, its complete removal appears to enhance its aggregation properties and its stability, whereas partial truncation has the opposite effect.
Next, we investigated whether the filamentous structures formed by the ∆N1-20 version of MyD88 were associated with any cellular membrane. To this end, we co-expressed it with fluorescent markers for the plasma membrane, ER and mitochondria (Fig. 2d). To assess plasma membrane association, we used the phosphatidylserine-binding domain Lact-C2 fused to mRFP (mRFP-Lact-C2), which labels the inner leaflet of the plasma membrane [36]. No association of MyD88-ΔN1-20 filaments with the plasma membrane was observed. For ER visualization, we employed the Sec63-mRFP marker. Although MyD88-∆N1-20 filaments did not co-localize with the ER, their ends frequently converged or were in close proximity to perinuclear ER regions (Fig. 2 d, arrowhead; see focal planes in Fig. S3). Co-localization analyses with the mitochondrial marker Ilv6-mCherry revealed that MyD88-ΔN1-20 filaments were often positioned near and longitudinally associated with yeast mitochondria. These findings suggest that MyD88-∆N1-20 filaments extend from ER-mitochondria contact sites, consistent with ERMES localization (see arrowhead in zoomed image in Fig. 2 d). Finally, co-expression of MyD88-ΔN1-20-EGFP with the Mdm34-DsRed marker confirmed co-localization with this ERMES component (Fig. 2e). Thus, the N-terminal 20-amino acid extension is dispensable for MyD88 attachment to ER-mitochondria contact sites.
Although our results suggest that the overlooked N-terminal extension of MyD88 seems to play an important role in its aggregation properties and its stability, AlphaFold2 did not predict significant structural differences between WT MyD88 and either its ∆N1-20 or ∆N1-7 versions (data not shown). However, interestingly, comparative structural modeling of oligomeric assemblies using AlphaFold2-multimer revealed notable differences between the predicted organization of four WT MyD88 molecules and four MyD88-∆N1-20 molecules. While MyD88 molecules appeared to nucleate around a single central point, MyD88-∆N1-20 molecules assembled in a filamentous fashion (Fig. 2f). According to the Predicted Aligned Error (PAE) graphs obtained (Fig. S4), the relative positioning of the TIR domains within these complexes is more precisely defined than that of the Death domains. This differential structural prediction of the intermolecular interactions may explain the distinct aggregation patterns observed in yeast, with WT MyD88 forming compact puncta and MyD88-∆N1-20 assembling into filaments.
Dynamin-like protein Dnm1 is necessary for proper MyD88 expression in yeastTo obtain genetic evidence supporting the association of MyD88 with ER-mitochondria contact sites, we attempted to construct yeast strains lacking individual ERMES components, specifically knocking out MDM34, MMM1, MDM10, or MDM12 genes. However, as previously reported [37], these deletions result in severe defects in mitochondrial DNA maintenance and inheritance. Consequently, the mutants acquired a petite phenotype, as has also been described by other authors [38]. In our hands, these mutants exhibited high clonal variability, preventing the consistent expression of the heterologous protein and yielding inconclusive results. Therefore, we opted to use a dnm1Δ deletion mutant, which lacks dynamin 1 (Dnm1) [39] and exhibits a less severe mitochondrial phenotype. Dnm1 plays a key role in mitochondrial fission, and its deletion leads to collapsed mitochondria [40]. Despite this alteration, ERMES integrity remains intact in dnm1Δ mutants, although the number of ER-mitochondria contact sites is reduced. Notably, mitochondrial inheritance is more efficient in dnm1Δ cells compared to ERMES mutants [41].
A strong loss of the amounts of MyD88 and MyD88-∆N1-20 was observed in the dnm1∆ mutant, as both proteins were detected at much lower levels compared to the WT strain, as assessed by fluorescence microscopy (Figs. 3a-c) and by Western blotting (Fig. 3 d). However, their subcellular localization pattern did not vary substantially in the dnm1∆ mutant as compared to the WT strain. Although the mitochondrial network was completely unstructured, in the few cells where MyD88-EGFP fluorescence was still detectable (approximately 2% for WT MyD88 and 10% for the truncated version; see Fig. 3c), full-length MyD88 remained adjacent to mitochondria (Fig. 3a), and MyD88-ΔN1-20 filaments also remained associated with them (Fig. 3b). These observations suggest that the ERMES continues to serve as a nucleation point for both constructs, but Dnm1 appears to be essential for maintaining levels of both MyD88 and MyD88-ΔN1-20 in the yeast cell.
Fig. 3
The expression of MyD88 and MyD88-∆N1-20 is reduced in a dnm1∆ mutant. (a, b) Maximum projection of Z-stack fluorescence microscopy images of BY4741 wild type (WT) cells (upper panels) and SEY5 (dnm1∆) mutant cells (lower panels), co-transformed with YEplac112-Ilv6-mCherry and pAG425-MyD88-EGFP (a) or pAG425-MyD88-ΔN1-20-EGFP (b). Scale bar corresponds to 5 μm. (c) Bar graph showing the percentage of cells with detectable MyD88-EGFP or MyD88-ΔN1-20-EGFP fluorescence in WT and dnm1Δ strains, as observed by fluorescence microscopy. The experiment was performed as a biological triplicate, with > 100 cells analyzed per transformant. Error bars represent SD. Asterisks indicate a p value < 0.01 (**), calculated using Student’s t test. (d). Immunoblot of cell extracts from BY4741 and isogenic dnm1∆ strains transformed with pAG425-MyD88-EGFP. MyD88 variants were detected using an anti-GFP antibody, with anti-G6PDH serving as a loading control. Quantification shows the ratio of each band’s intensity relative to the loading control, normalized to the corresponding construction in the WT strain
Linking the N-terminal localization domain of TIRAP to MyD88 shifts its affinity from plasma membrane to mitochondriaThe TIRAP adaptor consists of two main domains: the PtdIns(4,5)P2-binding domain (PBD), spanning amino acids 15–35 and responsible for plasma membrane localization [17], and the TIR domain, which interacts with MyD88 [42] (Fig. 4a). S. cerevisiae is a reference model organism in the study of phosphoinositide-dependent signaling, as its plasma membrane, like that of mammalian cells, is enriched in PtdIns(4,5)P₂ [43]. We have previously reported that human TIRAP is localized to the plasma membrane when expressed in yeast [24].
Fig. 4
Subcellular localization of TIRAP-MyD88 chimeric proteins. (a) Fluorescence microscopy images of YPH499 cells transformed with the plasmids pAG425-Ø-EGFP, pAG425-TIRAP-EGFP, pAG425-TIRAP-PBD-EGFP, pAG425-TIRAP-PBD-MyD88-∆N1-20-EGFP, pAG425-MyD88-EGFP, or pAG425-MyD88-N1-20-TIRAP-∆1-35-EGFP, as indicated in the schematic diagram on the left. Protein fragments in the chimeric constructs are linked through a glycine-alanine bridge (Gly-Ala×5). (b) Fluorescence microscopy images of YPH499 cells co-transformed with YEplac112-Ilv6-mCherry and pAG425-TIRAP-PBD-MyD88-∆N1-20-EGFP. Scale bar corresponds to 5 μm. All experiments were performed in biological triplicate, and representative images are shown
To investigate whether MyD88 localization could be redirected from the ERMES to the plasma membrane, we replaced its N-terminal extension with the PDB signal of TIRAP. As a control, we expressed a TIRAP-EGFP fusion that localized at the plasma membrane (Fig. 4a). In contrast to our previous observations reporting non-toxicity of TIRAP in yeast with another expression vector [24], yeast cells expressing TIRAP from the pAG425 plasmid exhibited moderate growth inhibition (Fig. S1c), likely due to the higher expression levels achieved (Fig. S5). The PBD domain alone was sufficient to drive EGFP to the plasma membrane (Fig. 4a) in 100% of the cells. However, the chimeric construct in which the first 20 amino acids of MyD88 were replaced by the PBD signal (TIRAP-PBD-MyD88-∆N1-20) did not decorate the plasma membrane. Instead, it was found in 96.26 ± 0.45% of the cells located in cytoplasmic organelles (Fig. 4a) that, upon co-expression with the Ilv6-mCherry marker, were identified as mitochondria (Fig. 4b). This chimera was expressed with a lower efficiency than WT MyD88 (Fig. S5), indicating that the addition of the TIRAP-PBD N-terminal extension was deleterious for MyD88 self-assembly.
Conversely, replacing the PDB domain of TIRAP with the N-terminal extension of MyD88 (MyD88-N1-20-TIRAP-Δ1–35) abolished its plasma membrane localization, resulting in a diffuse cytoplasmic distribution (Fig. 4a) in 98.55 ± 0.42% of the cells. This further supports the idea that no specific spatial signals lie in the first 20 amino acids of MyD88. Unlike WT TIRAP, none of these fusions or truncations were toxic for the yeast cell (Fig. S1c). Overall, these data suggest that the affinity of the MyD88 Death domain for mitochondrial membranes is dominant over the characteristic ability of TIRAP’s PDB to bind PtdIns(4,5)P2 at the plasma membrane.
The MyD88 TIR domain alone accumulates at lipid droplets, condensates mitochondria and is mildly toxic in S. cerevisiaeAfter determining that the MyD88 TIR domain is dispensable for its localization at yeast ERMES, we decided to study this domain in isolation. To this end, we developed two new MyD88 variants expressed from the same vector: MyD88-INT-TIR, encompassing amino acids 110–296, and MyD88-TIR, spanning amino acids 161–296 (Fig. 5a). These truncated MyD88 variants, which lack the Death domain, were immunodetected to a much greater extent than the wild type protein, suggesting increased protein stability in yeast (Fig. S5b). Notably, expression of MyD88-TIR, but not MyD88-INT-TIR, induced a certain degree of toxicity to the yeast cell (Fig. 5b).
Fig. 5
The TIR domain of MyD88 alone localizes to lipid droplets in yeast. (a) Schematic representation of MyD88 variants: wild type (MyD88), the INT and TIR domains (INT-TIR), and the isolated TIR domain (TIR). (b) Drop growth assay of YPH499 cells transformed with pAG425-Ø-EGFP, pAG425-MyD88-EGFP, pAG425-MyD88-INT-TIR-EGFP, or pAG425-MyD88-TIR-EGFP. (c) DIC and fluorescence microscopy images of YPH499 cells transformed with the plasmids listed in panel (b). The arrowhead indicates areas of enhanced refringency within the cells. Representative fields are shown. The graph at the right shows the percentage of cells with different kinds of MyD88 cytoplasmic spots, as noted. Error bars represent the SD from three different biological replicates (n≈50 for each transformant clone). Asterisks indicate a p-value < 0.001 (***), based on Student t-test analyses. (d) DIC and fluorescence microscopy images of YPH499 cells co-transformed with pGREG505-Erg6-mCherry and either pAG426-MyD88-TIR (top) or pAG426-MyD88-INT-TIR (bottom). (e) Expression of MyD88-TIR induces mitochondrial condensation in yeast. Representative DIC and fluorescence microscopy images of YPH499 cells transformed with the empty pAG425 plasmid or pAG425-MyD88-TIR. Scale bars correspond to 5 μm. (f) Ribbon and surface representations (top and bottom panels, respectively) of the three-dimensional structure of the MyD88 TIR domain (PDB: 4DOM). The BB loop (RDVLPGT), βE sheet, EE loop and αE helix are highlighted in yellow. Mutated residues are shown in color: magenta (R196C), blue (P200), and green (L252). (g) Loss-of-function mutations in the TIR domain eliminate toxicity in yeast. Drop growth assay of YPH499 cells transformed with plasmids pU323-Ø-Venus, pU323-MyD88-TIR-Venus, pU323-MyD88-TIR(R196C)-Venus, pU323-MyD88-TIR(P200H)-Venus, or pU323-MyD88-TIR(L252P)-Venus. Serial dilutions were plated on SD and SG-His synthetic media. All experiments were performed as biological triplicates; representative results are shown
The subcellular localization pattern of these variants remained punctate but differed from that of the wild-type protein (Fig. 5c). In most cells, the INT-TIR-EGFP variant formed a single large dot per cell, accompanied by smaller puncta scattered throughout the cytosol. On the other hand, TIR-EGFP predominantly formed small clustered dots in regions exhibiting refractive changes under differential interference contrast (DIC) microscopy (see arrowhead in Fig. 5c), suggesting an association with lipid droplets (LDs). This hypothesis was confirmed by co-expressing MyD88-TIR-EGFP with Erg6-mCherry, a marker of both LDs and the ER, as LDs originate from the ER and often remain associated with it [44] (Figs. 5 d, upper panels, and S6a). Consistently, MyD88-TIR-EGFP spots were also found adjacent to perinuclear ER regions marked with Sec63-RFP (Fig. S6b). However, in the presence of the INT domain, INT-TIR-EGFP did not co-localize with Erg6-mCherry spots (Fig. 5 d, lower panels). The precise identity of the compartments where MyD88-INT-TIR-EGFP localized was not further investigated, but we ruled out their association with the JUNQ and IPOD proteostatic stress compartments through co-expression with the markers ChFP-Ubc9ts and Rnq1-mCherry [45] (Fig. S6c).
Higher expression levels of MyD88-TIR, achieved by using a different expression vector with a Venus fluorescent protein fusion (Fig. S7b), resulted in a clearer association with ER membranes, particularly at the cell periphery. In some cases, this also lead to filamentous structures spanning through the cytoplasm, suggesting self-association (Fig. S7d). This implies that expression levels (or perhaps the use of different GFP variants) affect MyD88-TIR self-association and its interaction with cellular membranes. Additionally, we overexpressed the TIR domain of TIRAP, deprived of its N-terminal plasma membrane localization signal, as a Venus fusion protein (Fig. S7c). As expected, this TIRAP-TIR-Venus fusion failed to localize to the plasma membrane and, like MyD88-TIR, localized to cytoplasmic spots coincident with areas of refringency changes in transmitted light microscopy (Fig. S7e). Taken together, these findings indicate that isolated TIR domains from human TLR signaling adaptors, when detached from their native localization signals, have an intrinsic affinity for yeast lipid droplets.
The observation of mitochondria using the Ilv6-mCherry marker in MyD88-TIR-EGFP-expressing cells revealed defects in mitochondrial morphogenesis. Mitochondria frequently appeared condensed near one of the cell poles, where the MyD88-TIR-EGFP signal was found in close proximity, overlapping with regions of refractive change in DIC microscopy (Fig. 5e, see arrowhead). These findings suggest that overexpression of the isolated MyD88 TIR domain disrupts ER-associated membranes, leading to mitochondrial dysfunction. This disruption may underlie the yeast growth inhibition observed specifically in this truncated MyD88 variant.
To determine whether TIR-TIR self-interaction was involved in the toxicity observed, we introduced point mutations in the asymmetric interaction interfaces of the MyD88 TIR domain, specifically in the BB loop and the βE/EE/αE loop, which are critical to homopolymer formation (Fig. 5f) [46, 47]. We generated the BB loop loss-of-function mutations R196C and P200H, the latter being equivalent to the P125H mutation within the TIR domain of TIRAP, which prevents TIR-TIR interactions [2, 7]. As a third variant, we introduced the oncogenic L252P mutation [48], which affects a residue located in the βD sheet, a region that is considerably less exposed (Fig. 5f). Loss-of-function mutants R196C and P200H no longer induced the slight growth inhibition observed with the wild-type TIR domain, whereas the gain-of-function mutant L252P exhibited a behavior similar to that of the wild type in this experimental setting (Fig. 5 g). Growth recovery in the loss-of-function mutants was not due to decreased protein stability, as immunoblot analyses confirmed that mutant protein levels were equivalent to those of the wild type version (Fig. S8a). Finally, fluorescence microscopy revealed that MyD88-TIR-Venus R196C, P200H, and L252P point mutants failed to form cytoplasmic filaments. Instead, all three mutants retained ER localization but tended to form compact spots (Fig. S8b).
A sensitive fluorescence assay to test MyD88 interactions in vivo: the pathological L252P mutation limits self-associationAccumulation of MyD88 at the ERMES or other cytoplasmic membranes could reflect accumulation of the misfolded heterologous protein. To rule out this possibility, we aimed to develop a yeast-based system capable of quantitatively assessing physiological MyD88 self-interactions. For this purpose, we used the tripartite GFP system [49] adapted to yeast [30]. Briefly, this system relies on the fusion of GFP β10 and β11 sheets to two putative interacting proteins, enabling the reconstitution of GFP over an incomplete β(1–9) GFP barrel upon interaction (Fig. 6a). We hypothesized that if properly folded MyD88 molecules were fused to both GFP β10 and β11 sheets, their polymerization would bring the sheets into proximity, leading to GFP reconstitution at the ERMES. To test this, we fused each β sheet to MyD88 at its C-terminus and studied self-interaction by these means. Control experiments using the individual fusions with plasmids expressing only the 3xFLAG-6xHIS tag fused to the corresponding β-sheet did not produce any fluorescence signal (data not shown), whereas the co-expression of MyD88-β10 and MyD88-β11 resulted in a robust fluorescent signal, indicating successful reconstitution of the tripartite GFP protein (Figs. 6c, d).
Fig. 6
The oncogenic L252P mutation in MyD88 limits oligomer formation in yeast. (a) Schematic representation of the tripartite GFP assay. Two plasmids expressing the respective fusions of β10 and β11 GFP sheets to each of the proteins of interest are transformed into a yeast strain expressing a truncated incomplete β(1–9) form of GFP. Fluorescence is reconstituted only when the two proteins physically interact in space and time. (b) Diagram of human chromosome 3 showing the pathological point mutation that substitutes leucine 252 with proline in the MyD88 protein. (c) DIC and fluorescence microscopy images of AF1 strain cells transformed with plasmids pU316-3xFLAG-6xHis-MyD88-β10 and pU313-3xFLAG-6xHis-MyD88-β11 (left panels), or pU316-3xFLAG-6xHis-MyD88(L252P)-β10 and pU313-3xFLAG-6xHis-MyD88(L252P)-β11 (right panels). Experiments were performed as a biological triplicate, and representative images are shown. The scale bar corresponds to 5 μm. (d) Flow cytometry analysis. The left panel shows overlaid histograms where GFP fluorescence intensity is plotted on the X-axis and the number of events on the Y-axis. A representative result of a biological triplicate is shown. The right panel presents a bar graph of the mean fluorescence intensity of each sample. Error bars represent the SD. Statistical significance was determined using Tukey’s HSD test (***p < 0.001)
To evaluate the performance of the tripartite GFP assay, we examined the effect of the oncogenic L252P mutation on MyD88 self-interaction. This variant, reported as constitutively active, is the most prevalent mutation in Waldenström macroglobulinemia (Fig. 6c) [50]. Fluorescence microscopy revealed that this purported gain-of-function mutant formed a similar number of cytosolic puncta per cell as the wild type protein but with significantly lower fluorescence intensity (Fig. 6b). Western blot analyses confirmed that this reduction was not due to differences in protein expression levels (Fig. S9). As shown in Fig. 6d, this assay provides quantifiable data via flow cytometry. The difference in mean fluorescence intensity (MFI) between the wild type and L252P samples was statistically significant (Fig. 6 d, right panel). To sum-up, this yeast-based system represents a valuable tool for genetic and pharmacological screening of mutations or compounds that modulate MyD88 interactions.
Comments (0)