
Remember me

Murine-derived breast cancer cell line 4T-1 was purchased from the Chinese Academy of Sciences Cell Bank (Shanghai, China) and were cultured in RPMI medium (Gibco BRL) supplemented with 10% fetal bovine serum, penicillin, and streptomycin (50 U/mL). All cultures were maintained at 37 °C in a 5% CO₂ atmosphere.
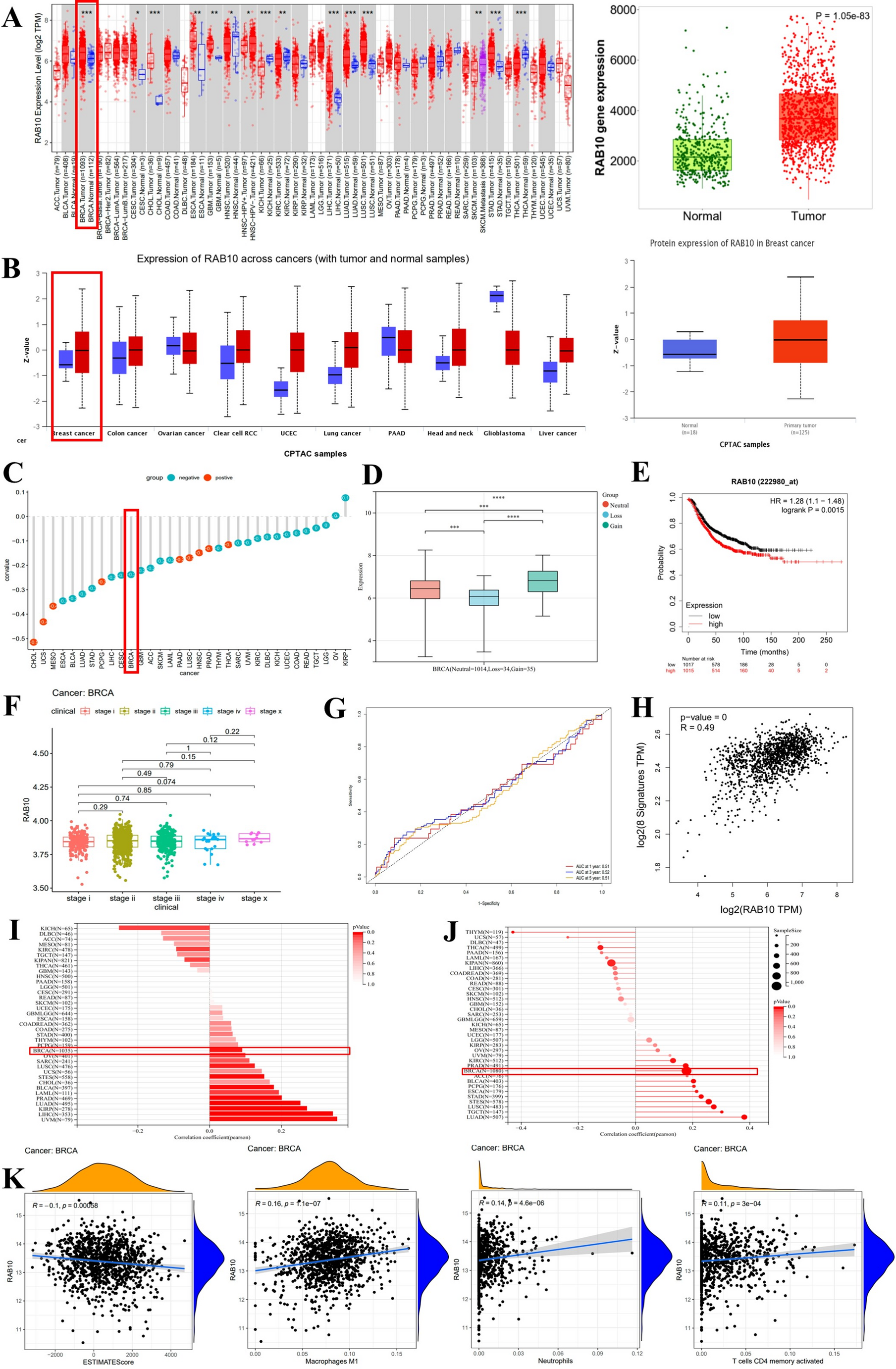
Online data acquisition and analysesRNA sequencing data of BC patients were obtained from The Cancer Genome Atlas (TCGA) database. Differential expression analysis for the RAB10 gene between tumors and adjacent normal tissues in the TCGA database was conducted using the “Exploration-Gene DE” module of the TIMER2.0 network server [6]. The UALCAN database was used to access data from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) and the International Cancer Proteogenome Consortium (ICPC) datasets for pan-cancer analysis of RAB10 protein expression, as well as for differential expression analyses between BC and normal tissue [7]. To assess the relationship between RAB10 expression and DNA methylation within the TCGA database, we utilized the SMART platform to provide a comprehensive analysis of DNA methylation in the TCGA project [8]. The correlation between RAB10 expression and tumor stage was analyzed using the R packages “limma” and “ggpubr”. Survival analysis using gene chip data was performed with Kaplan–Meier Plotter to generate a survival prognostic model of RAB10 in BC [9]. The R package “pROC” was used to analyze the ROC curve correlation index of RAB10 expression in BC patients. The TIMER 2.0 database [6] “immune-gene” module, with purity adjustment, was utilized to investigate the association between RAB10 expression and tumor-infiltrating immune cells (TIICs) in BC. The SangerBox tool was used to assess the RNAss, LOH, and CNV multiple indicators for RAB10. The “Correlation Analysis” function of GEIPA2 and RAB10 was used to analyze the correlation of several genes (SLC2A2, HK2, PFK1, SIRT1, G6P, LDHA, FOXO1, DGAT1, GLUT1).
The construction of RAB10-deficient mouse model for breast cancerA lentiviral vector (LV-shR) containing the RAB10 interference fragment was constructed and transfected into the murine breast cancer cell line 4T-1. After 72 h, 10 µg/µL of puromycin was added for selection to obtain mouse breast cancer cells stably expressing RAB10 deficiency (4T-1/sh-RAB10). BALB/c female mice were divided into two groups: a negative control group and a 4T-1/sh-RAB10 group, with at least eight mice in each group. A subcutaneous tumor model of mouse breast cancer was established. The tumor size adhered to medical ethical standards. Mice were sacrificed after 3 weeks, and tumor tissues were harvested for proteomic and metabolomic analysis as well as immunohistochemical staining.
Proteomic LC- MS analysis (TMT10plex) and data analysisTotal proteins were extracted from tumor tissues. We adopted a method combining liquid nitrogen grinding and chemical lysis: The tumor tissue was rapidly ground into powder in liquid nitrogen, and then RIPA lysis buffer containing protease inhibitor and phosphatase inhibitor was added. The tissue was lysed on ice for 30 min, with vortex oscillation three times, each time for 10 s. The lysis buffer was centrifuged at 14,000 × g for 15 min at a temperature of 4 °C. Extract the supernatant, determine the concentration using the BCA protein quantification kit, and adjust it to a uniform concentration (1–2 mg/mL) for subsequent experiments. In this study, 8 biological replicates were set up in the proteomics experiment (tumor tissues were extracted independently from 8 mice in each group), and 6 analytical replicates (i.e., the same sample was injected 6 times in parallel) were performed for each biological replicate sample to ensure the stability and reliability of the data. Data from the analytical replicates were used to calculate the coefficient of variation (CV), and only data with a CV of less than 15% were retained for final analysis. The resulting LC-MS/MS raw files were imported into Proteome Discoverer 2.4 software for data inter pretation and protein identification using the Sequest HT database to obtain differentially expressed proteins. Bioinformatics data were analyzed using Perseus software, Microsoft Excel, and R statistical computing software. Biological replicates were included in this experiment, and cut-off values for significant differential expression were set at a ratio fold-change > 1.20 or < 0.83 and P < 0.05. Subsequently, GO/KEGG enrichment analysis was performed by clusterProfiler package, PPI network was constructed by combining with STRING database and visualized by Cytoscape, and finally, the data were visualized by GraphPad Prism and R language ggplot2, which systematically revealed the protein function and pathway changes associated with RAB10 knockdown.
A modified FASP (filter-assisted sample preparation) method was used. Extracted protein solutions were processed in 10 kDa ultrafiltration tubes, then reduced (10 mM DTT, 56 ℃, 30 min), alkylated (55 mM iodoacetamide, room temperature, 20 min), and digested with trypsin (enzyme-to-substrate ratio of 1:50, 37 ℃, 16 h), and then the peptides were lyophilized and set aside for final analysis. Mass spectrometer and setup: A Thermo Scientific Q Exactive HF-X mass spectrometer and an Easy nLC 1200 liquid chromatography system were used. Chromatographic conditions were as follows: C18 reversed-phase column (75 μm × 150 mm, 2 μm, 100 Å), mobile phases A (0.1% formic acid aqueous solution) and B (0.1% formic acid acetonitrile solution), gradient elution (5–35% B, 90 min).
Data normalizationThe median normalization method was used to globally normalize all samples labeled with TMT to eliminate systematic errors among different samples.
Handling of missing valuesProteins with a percentage of missing values below 10% were interpolated using the K-Nearest Neighbor algorithm (KNN); if the missing values exceeded 10%, they were excluded from the analysis.
FDR controlThe identified peptides and proteins are validated using the Percolator algorithm to control the false discovery rate (FDR) to ≤ 1%, and each protein is required to contain at least 2 unique peptides to ensure its reliability.
Metabolomic UHPLC- MS/MS analysis and data analysisMetabolite extraction was performed by grinding the tissue, and the supernatant was processed using a tissue crusher for mass spectrometry analysis. Metabolomics analysis was carried out using a UPLC-ESI-Q-Orbitrap-MS system coupled with a Q-Exactive Plus mass spectrometer. Metabolites with variable importance in projection (VIP) values > 1.0 and P < 0.05 were considered statistically significant. KEGG enrichment analyses were conducted using Fisher’s exact test, and FDR correction for multiple testing was applied. Enriched KEGG pathways were considered nominally significant at P < 0.05.
It should be explained that the core difference between “grinding” and “tissue crusher” in metabolomics analysis lies in the crushing principle and the applicable scenarios: the former grinds tissues into powder by mechanical friction (e.g., mortar + liquid nitrogen, oscillation of beads) and relies on cryogenic environment (e.g., liquid nitrogen cryomilling) to inhibit enzyme activity to protect metabolites. The former grinds the tissue into powder by mechanical friction (e.g. mortar + liquid nitrogen, oscillation of grinding beads), and relies on low temperature environment (e.g. liquid nitrogen freezing and grinding) to inhibit enzyme activity to protect metabolites, which is suitable for small-volume, brittle tissues and polar metabolite analysis; the latter utilizes high-speed mechanical shear force to crush, which is suitable for large-volume, tough tissues or high-throughput processing, but it may affect the stability of heat-sensitive metabolites due to the heat generation. Therefore, in this study, liquid nitrogen milling was adopted for tumor tissues to inhibit the endogenous enzyme activity of tissues through low-temperature mechanical crushing, avoiding metabolite degradation during the crushing process and ensuring the efficient release of intracellular polar metabolites, which is suitable for the precise analysis of complex metabolomes in the tumor microenvironment.
RNA extraction and qRT-PCRBreast cancer cells were collected by trypsin digestion and lysed using a Trizol extraction kit (Invitrogen, Carlsbad, CA) to isolate total RNA. Reverse transcription of total RNA into cDNA was performed using a reverse transcription kit (Thermo Scientific, USA). cDNA was amplified by polymerase chain reaction (PCR) using a qRT-PCR kit (GeneCopoeia, Guangzhou, China). Primer sequences used are shown in Table 1. The relative expression of target genes between different groups was calculated using the relative quantification algorithm ΔΔCt method.
Table 1 Primers for RAB10 and GAPDHWestern blotMouse breast cancer 4T-1 cells were collected and lysed on ice for 30 min using RIPA buffer with PMSF (RIPA = 99:1) to prepare the protein lysis solution. The supernatant was collected by centrifugation at 12,000 rpm for 10 min at 4 °C. Total protein concentration was measured using the BCA Protein Assay Kit (Pierce). After several pre-tests, a sample volume of 30 µg total protein per well was used. Nitrocellulose membranes were blocked in blocking solution for 2 h at room temperature, followed by overnight incubation with the primary antibody at 4 °C. The membrane was then washed with TBST buffer for 30 min, incubated with the secondary antibody for 2 h at room temperature, and washed again for 30 min. A developer solution was prepared for exposure and development.
The details of the gel ingredients:
1.Lower gel (separating gel) systems
Acrylamide/Bisacrylamide (Acr-Bis):
Provides premixed solutions at different concentrations to form a gel matrix for separation of proteins by molecular weight.
Tris-HCl buffer (2×):
Maintains the pH environment of the separation gel (typically 8.8, consistent with conventional SDS-PAGE) and contains SDS (sodium dodecyl sulfate) to denature and negatively charge the proteins, ensuring that electrophoretic mobility is only molecular weight dependent.
Improved coagulant promoter:
Replaces traditional APS (ammonium persulfate) and TEMED (tetramethylethylenediamine) with a stable catalytic system for rapid initiation of gel polymerization at room temperature (15–30 min) without the need to add additional TEMED and to avoid off-odors.
2.Upper Gel (Concentrated Gel) Systems
Low concentration of acrylamide/bisacrylamide:
Typically 4.2%, used to concentrate protein samples at the beginning of electrophoresis to improve band resolution.
Color Buffer (2×):
Contains Tris-HCl buffer (pH 6.8, presumably) and red, blue, and green dyes to facilitate differentiation of different gels during spotting and to provide visual markers without affecting subsequent staining or membrane transfer.
Specific primary and secondary antibodies are shown in Table 2:
Table 2 Source of antibodies and secondary antibodiesWound healing assayCell migration was assessed using a wound healing assay. Cells were plated in 6-well plates (3 × 10⁵ cells/well) and incubated in RPMI-1640 medium (Hyclone; GE Healthcare) without FBS at 37 °C until reaching 80% confluency. A scratch was made across the surface of the well using a 10-µL pipette tip. Scratches were observed at 24 and 48 h after incubation at 37 °C.
Cell proliferation assayThe effects of different treatment groups on the viability of 4T-1 cells were evaluated using the CCK-8 assay (Two groups: the control group and the gene knockdown RAB10 group; Three groups: the control group, the gene knockdown Slc37a2 group, and the gene knockdown Slc37a2 combined with mTOR activator MHY1485 group). Cells were uniformly seeded in 96-well plates at a density of 5 × 10³ cells per well in 200 µL of 1640 medium and incubated at 37 °C for 24, 48, and 72 h. After adding 10 µL/well of CCK-8 reagent (Biosharp, China), cells were incubated for 1–2 h at 37 °C. Cell viability was measured by reading the absorbance at 450 nm using a microplate reader (Thermo Scientific, USA). The effects of different treatment groups on the proliferative ability of 4T-1 cells were also assessed using a cell colony formation assay. Cells were uniformly seeded in six-well plates at a density of 500 cells per well, with medium changes every 3–4 days. Clones formed within 10–15 days, were fixed with paraformaldehyde, stained with crystal violet, photographed, and recorded.
Transwell assaysTranswell assays were performed in a 24-well Transwell unit with an 8-µm pore-size polycarbonate membrane. Matrigel (100 µL; 300 µg/mL) was added to the upper wells. Cells that had migrated to the lower membrane surface were stained with crystal violet and counted in five high magnification fields (400×). All samples were tested in triplicate, and the experiment was repeated three times.
In this study, cellular experiments (e.g., CCK-8, scratch, plate clonogenic assay, Transwell, Western Blot, etc.) were performed with 3 biological replicates and corresponding technical replicates (e.g., 3 replicate wells/group), with the same operating parameters such as cell density, treatment time, and reagent concentration to ensure comparable results.
Immunohistochemistry (IHC) and TUNEL staining analysisMouse breast cancer tissue was fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned to a thickness of approximately 3 μm. Tumor tissue-associated antibodies were detected by overnight incubation at 4 ℃, followed by washing with PBS, incubation with secondary antibody (green fluorescence) for 2 h, restaining with DAPI, washing with PBS, and mounting with glycerol for IHC index evaluation. TUNEL apoptosis was detected using the TUNEL Apoptosis Detection Kit (Servicebio). Each section was treated with 50 µL of detection solution, incubated at 37 °C for 60 min, and washed with PBS. The primary antibody used was RAB10 (Abclonal, A22746), p62(Proteintech, 18420-1-AP), LC3 (Proteintech, 14600-1-AP), HK2 (Proteintech, 22029-1-AP), GLUT1(Abclonal, A6982), E-cadherin (Proteintech, A11509), snail (Proteintech), CD206 (Proteintech, 18704-1-AP), Arg-1(Cell Signaling Technology, 93668), PCNA (Proteintech, 10205-2-AP). Secondary antibody using Goat Anti-Rabbit IgG(H + L) FITC -conjugated(Affinity).
Immunofluorescence assayCells were seeded into confocal dishes (Biosharp, BS-20-GJM) at a density of 2 × 10⁴ cells per well. Cells were fixed with 4% paraformaldehyde for 20 min and incubated with the primary antibody overnight at 4 °C, followed by incubation with a fluorescent secondary antibody (1:200) for 1 h at room temperature. Subsequently, cells were stained with DAPI (1:1000) for 10–15 min in the dark and observed under a laser scanning confocal microscope.
ApoptosisWe performed apoptosis experiments using the Annexin V-FITC/PI Double Staining Apoptosis Detection Kit (Bestbio, BB-4101). 4T-1 cells were inoculated in 6-well plates at 5 × 10⁴ cells/well and divided into three groups: (1) control group; (2) si-Slc37a2 group (transfected with 50 nM Slc37a2 siRNA); and (3) si-Slc37a2 + MHY1485 group (transfected with Slc37a2 siRNA for 24 h and then added 5 µM Cells were collected after 24 h of MHY1485 treatment, rinsed with PBS and resuspended in 100 µL of binding buffer, then 5 µL of Annexin V-FITC and 5 µL of PI were added, and incubated for 15 min under low light conditions before apoptosis analysis was performed on a BECKMAN Coulter flow cytometer. The stains used were Annexin V-FITC (fluorescein isothiocyanate-labeled membrane-bound protein V) and PI (propidium iodide), both from the Annexin V-FITC/PI kit.
Glucose consumption, lactate, G6P, citrate (CA), ATP, lipid Peroxidation (LPO) and Reactive Oxygen Species (ROS) Generation, mitochondrial membrane potential (MMP) and GSH activity assaysCells were seeded into six-well plates at a density of 1 × 10⁶ cells in 2 mL RPMI 1640 medium per well and cultured overnight. Glucose consumption, lactate production, G6P production, citrate production, ATP production, ROS generation, LPO production, and mitochondrial membrane potential were detected using the following kits: glucose assay kit (Nanjing Jiancheng Bioengineering Institute A154-2-1), LD assay kit (Nanjing Jiancheng Bioengineering Institute A019-2-1), G6P content assay kit (Beyotime S0815), citric acid (CA) content assay (Solarbio® BC2150), ATP content assay kit (Nanjing Jiancheng Bioengineering Institute A095-2-1), ROS content kit (Beyotime S0033S), LPO kit (Thermo Scientific C10445), mitochondrial membrane potential detection kit (JC-1) (Beyotime C2006), and GSH kit (Solarbio® BC1175), according to the manufacturers’ instructions. All experiments were performed at least three times, and data were normalized by cell numbers or protein content.
Specific experimental details:
1.Experiments in glucose metabolism
1.1Determination of cellular glucose consumption
(1)Preparation of working solution: The ratio of reagent R1 and reagent R2 is 4:1;
(2)Standard curve: Dilute 10 mM glucose standard with ddH2O to 1000 μM, 500 μM, 250 μM and 125 μM respectively, and use the sample containing only ddH2O as the control to draw the standard curve;
(3)Sample preparation: The cell density of each well was about 2×105 cells. After different treatments, the culture medium was collected at the specified time points, and the supernatant of the cells was aspirated after centrifugation at 1000 rpm for 10 min;
(4)Detection: The total volume of the reaction system in each well of the 96-well plate was 200 μL, and 3 replicate wells were set up for each sample. The sample addition procedure for each well is shown in Table 3:
Table 3 Standardized sample loading for cellular glucose detection (5)Incubate at 37℃ for 10 min, and measure the absorbance value of each well using an enzyme meter;
(6)Concentration analysis: The formula for calculating glucose concentration (mmol/L) is: concentration of standard solution (5.55 mmol/L) × sample dilution × (sample assay value - blank tube control value)/(standard tube assay value - blank tube control value).
1.2Determination of cellular lactate production
(1)Preparation of working solution: Mix Reagent II (enzyme reserve solution) with Reagent I (enzyme dilution solution) at a ratio of 1:100, taking care to prepare freshly before use;
(2)Sample preparation: the same as the experiment for the determination of glucose consumption;
(3)Take 1.5 mL EP tubes and label them according to different groups, add 250 μL of working solution to each EP tube, and then add the samples according to the Table 4 below:
Table 4 Standardized sample loading for cellular lactate detection (4)Add 50 μL of color development solution to each tube, shake and mix well, then incubate at 37℃ for 10 min, then add 500 μL of termination solution to each tube in turn, and vortex and shake;
(5)96-well plate assay: 200 μL of sample per well in a 96-well plate, and the absorbance value was detected by an enzyme labeling instrument;
(6)Concentration determination: Calculation formula for lactate concentration (mmol/L): (sample group assay value - blank group assay value) / (standard group assay value - blank group assay value) × standard solution concentration (3 mmol/L)
1.3Determination of cellular glucose-6-phosphate (G6P) production
(1)Sample preparation: The density of cells in each well was about 1×106. After removing the medium, cells were washed with PBS, then 200μL of G6P extract pre-cooled to ice was added and gently blown, and the whole crushing process was completed on ice. Subsequently, centrifuge at 12000g for 10 min at 4ºC, and take the supernatant as the test sample, or store at -20°C for spare;
(2)Preparation of G6P standard solution: Set the concentration range of 0, 31.25, 62.5, 125, 250, 500 μM, and use the G6P extract to dilute the G6P standard solution for detection. Add 50 μL of standard solution in each well during detection, where 0 μM concentration group is the blank control;
(3)Preparation of G6P detection solution;
(4)96-well plate assay: Transfer 50 μL of the test sample to a 96-well plate, set up 3 replicate wells for each group, add 50 μL of G6P assay solution to each well, mix gently to prevent air bubbles from being generated. incubate at 37ºC, avoiding light for 15-30min, when the color is orange-yellow, and the absorbance value is detected by the enzyme meter at 450 nm;
(5)Determination of G6P content: Plot the calibration curve, where the horizontal coordinate is the concentration of G6P and the vertical coordinate is the value of optical density. Subtract the value of the optical density of the standard and the sample from the value of the blank control, and calculate the concentration of G6P in the sample according to the calibration curve.
1.4Determination of cellular citric acid production
(1)Sample treatment: the cell density of each well is about 1×106, process the cells to the centrifuge tube for centrifugal separation and remove the supernatant. Add appropriate amount of reagent I in the centrifuge tube, operate on ice to sonicate the cells for lysis (sonicate 3s every 10s interval, repeat 30 times), then centrifuge at 10,000 g for 10 min at 4℃, and collect the supernatant on ice for testing;
(2)Preheat the spectrophotometer at least 30 min in advance, set the wavelength to 545 nm, and calibrate the zero point with ddH2O. Place reagent I in a 30°C water bath for more than 30 min. Add the appropriate reagents to the centrifuge tube according to the following steps;
(3)After sufficient mixing, incubate for 30 min at room temperature and measure the optical density at 545 nm;
(4)Calculation of citric acid concentration: Citric acid concentration (μmol/104 cell) = C standard solution × (A assay tube - A blank tube) / (A standard tube - A blank tube) × V total/number of cells.
2.Detection method of reactive oxygen species (ROS) and lipid peroxides (LPO)
(1)Sample treatment: Cells were spread in a six-well plate at a density of about 3×105 per well, cells were extracted at the specified time after different treatments, and the precipitate was collected after centrifugation;
(2)Probe loading: Dilute ROS probe and LPO probe according to 1:1000 to make the final concentration of 10 μmol/L. In situ probe loading: firstly, remove the cell culture solution, add 1-2 mL of diluted probe solution to each well, and after incubation for 30 min in 37ºC environment, wash the cells using serum-free culture solution with the purpose of removing unbound probes. Probe loading after cell extraction: the collected cell precipitate was resuspended with probe solution and placed in a 37ºC incubator for 30 min for reaction, during which time the cells were gently mixed every 5 min and washed using serum-free culture medium;
(3)Detection and analysis: The treated samples were examined using laser confocal microscopy or flow cytometry to quantify the levels of ROS and LPO.
3.Mitochondrial membrane potential assay
(1)Preparation of JC-1 staining working solution: Dilute JC-1 (200×) with ultrapure water at the ratio of 1:160 and vortex and oscillate to dissolve completely, then add buffer to obtain JC-1 staining working solution;
(2)Setting of positive control group: 10mM CCCP was added to the cell culture medium at a ratio of 1:1000 and reacted at 37ºC for 20 min, followed by JC-1 probe labeling synchronously with other treatment groups;
(3)JC-1 detection step: After discarding the culture medium and washing with PBS, add 1 mL of fresh culture medium to each well, and add 1 mL of JC-1 staining working solution to each treatment group, mix gently. The reaction was carried out in a thermostat at 37ºC for about 20 min. 5× of JC-1 staining buffer and distilled water were mixed in a 1:4 ratio to make 1× buffer and pre-cooled in an ice bath in advance. When the reaction was finished the cells were washed twice using 1× buffer. Finally, 2 ml of culture solution was added and observed and analyzed using fluorescence microscope or confocal microscope.
4.ATP assay
(1)Sample preparation: After the cells are spread on the plate and subjected to different treatments, remove the culture medium from the wells after they have proliferated to a suitable density, add 200 μL of lysate to each well, blow gently, and centrifuge the cells for 5 min at 12,000g under the condition of 4ºC, and collect the supernatant for the subsequent analysis;
(2)Establishment of standard curve: Dilute the ATP standard solution with concentrations of 0.01, 0.03, 0.1, 0.3, 1, 3 and 10 µM respectively, and measure the corresponding signal values according to these dilutions, and plot the standard curve for subsequent quantitative analysis;
(3)ATP assay preparation: Mix ATP assay reagent with ATP diluent at a ratio of 1:9;
(4)Determination of ATP concentration: 100 μL of ATP detection solution was added to the wells and allowed to stand for 5 min, followed by the addition of 20 μL of the sample or standard to be tested and mixing, and then the signal value was detected by chemiluminescence instrument. The actual concentration of ATP in the sample should be calculated according to the standard curve.
Comments (0)