Animals
Sprague Dawley (SD) rats (6 weeks, male, 180–220 g) purchased from Shanghai SIPPR-Bk Lab Animal Co., Ltd. They were maintained in specific pathogen-free (SPF) laboratory animal facilities of the Ninth People's Hospital Affiliated to Shanghai Jiao Tong University School of Medicine. All animal experiments complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and received approval from the Institution of Animal Care and Use Committee (IACUC) of the Ninth People’s Hospital Affiliated to Shanghai Jiao Tong University School of Medicine. We kept rats in individual cages under controlled conditions: a consistent temperature of 22 ± 2℃ and a 12-h light–dark cycle, with unrestricted access to water and food.
Rat PTJC model establishment
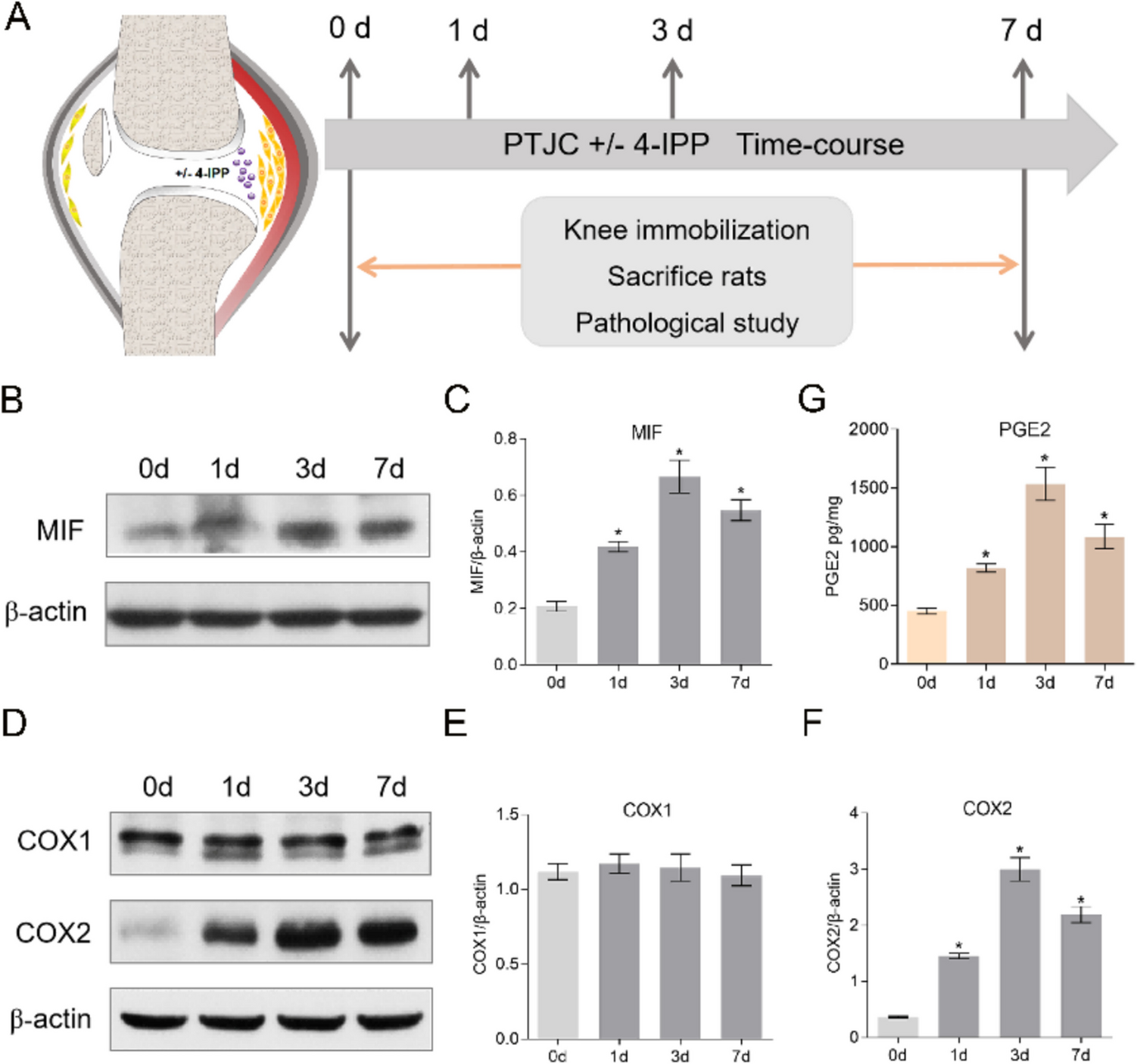
PTJC model establishment was referred to the foundation of previous study [28]. Briefly, six rats in each experimental group received an intraperitoneal injection of 50 mg/kg pentobarbital sodium for anesthesia and were placed supine for the procedure. The right knee joint area was shaved the fur and sterilized with chlorhexidine before making a 15 mm midline incision on the right hind leg’s posterior side. The patella was shifted medially to reveal the femoral condyles, allowing for the creation of two 1.5 × 1.5 mm cortical openings in the lateral and medial femoral condyles’ non-articular cartilage. The cruciate ligament was cut and the knee hyperextended to –45° to tear the posterior joint capsule. The knee was immobilized at approximately 135° of flexion using a 0.5-mm steel wire. The patellofemoral joint was then realigned, and closure was achieved with silk sutures. For drug administration, vehicle or 100 mM 4-IPP (TOCRIS, Bristol, UK) were injected into the knee joint cavity. Post-surgery, the sodium salicylate (150 mg/kg) was used for pain control and the rats freely moved in their cages until euthanasia on days 0, 1, 3, and 7. The knee joint and posterior capsule were harvested for subsequent assays.
Joint capsule fibroblasts isolation and culture
Based on prior studies [28], 4-week-old male SD rats were euthanized through overdose anesthesia and sterilized in 75% ethanol for 5 min. The posterior joint capsule tissue was obtained and washed in Dulbecco's minimum essential medium (DMEM; HyClone, Logan, USA) and cut into 2–3 mm pieces. These tissue pieces were transferred to DMEM containing 1% penicillin/streptomycin (Gibco, Grand Island, NY, USA) and 10% fetal bovine serum (FBS; Gibco), and cultured in a 37 °C, 5% CO2 environment. Joint capsule fibroblasts typically began migrating from the tissue pieces within 3–5 days. When cell coverage reached approximately 90%, cells were passaged to the second to fourth generation before used in subsequent experiments.
Small interfering RNA (siRNA) transfection
Once primary joint capsule fibroblasts reached a density of 40%−50%, siRNA-mediated gene silencing experiments were conducted. Specifically, 5 μL of Lipofectamine 3000 (Invitrogen) transfection reagent was pre-mixed with 250 μL of Opti-MEM. Separately, 5 μL of 20 μM CD74 siRNA2 (RiboBio, Guangzhou, China) or negative control siRNA were also diluted in 250 μL of Opti-MEM. Afterward, these solutions were combined and allowed to form complexes for 15 min. Then, the resulting complexes were added to 6-well plate containing 1500 μL of DMEM, gently mixed, and incubated in a 37 °C, 5% CO2 environment for approximately 48 h to permit gene silencing effects. CREB gene silencing (RiboBio, Guangzhou, China) was similarly performed using different siRNA sequences (siRNA1, siRNA2, siRNA3) following the same procedures.
Western blot
The samples from rat posterior joint capsules or joint capsule fibroblasts were collected and lysed using RIPA Lysis Buffer (Beyotime, Shanghai, China). Western blot was performed according to previous methods and protein band intensity normalized against β-actin expression [27]. Primary antibodies included β-actin (ProteinTech, Wuhan, 1:5000), MIF, COX1 (Abcam,Cambridge, UK, 1:1000); COX2 (Cayman, Michigan, USA, 1:500); CD74 (Santa Cruz, CA, 1:1000), p-CREB, CREB, TGF-β1, α-SMA (Abcam, 1:1000). Secondary antibodies were goat anti-rabbit IgG or goat anti-mouse IgG (Invitrogen, 1:2000). The Odyssey imaging system (Li-Cor, Lincoln, NE, USA) was used to determine fluorescent signals.
Quantitative real-time polymerase chain reaction (qRT-PCR)
RNA was extracted from joint capsule fibroblast or macrophage treated with MIF (ProSpec, USA), PGE2 (Sigma, MO, USA), siRNA (RiboBio, Guangzhou, China), or inhibitors using the TRIzol (Invitrogen) protocol. Total RNA was converted into cDNA using the Omniscript reverse transcription kit (QIAGEN). cDNA diluted 1:5 was used for qRT-PCR. Subsequently, qRT-PCR was conducted on Applied Biosystems real-time PCR system, utilizing Takara Bio’s SYBR® Premix Ex Taq™ (Takara Bio, Dalian, China). Briefly, 4 μL SYBR Green (G3320, Servicebio), 1 μL forward and reverse primers, 2 μL cDNA template, and 3 μL ddH2O were added to make a reaction volume of 10 μL for qRT-PCR analysis of the target gene. The primers were synthesized by Sangon Biotech (Shanghai, China) as Table S1. Gene relative expression levels were calculated using the 2-ΔΔCt method and normalized to Gapdh. Each reaction was performed in triplicate.
Immunofluorescence
For cells: Following MIF and 4-IPP treatment, joint capsule fibroblasts were fixed in 4% paraformaldehyde containing 0.1% Triton X-100 (Sigma, MO, USA) and 4% goat serum was used to block non-specific binding. Subsequently, joint capsule fibroblasts incubated with primary antibodies against COX2 (Cayman, 1:100), phalloidin (Abcam, 1:1000), and vimentin (Abcam, 1:1000) overnight.
For tissue: Rat knee joint specimens were fixed in 4% paraformaldehyde, decalcified in 10% EDTA, embedded in paraffin, and then sagittally sectioned (4 μm) using a microtome. The sections co-incubated with primary antibodies against MIF (Abcam, 1:100), vimentin (Abcam, 1:1000), COX2 (Cayman, 1:100), and CD74 (Santa Cruz, 1:50) overnight.
FITC-labeled goat anti-rabbit IgG (Sigma, 1:400) and CY3-labeled goat anti-rabbit IgG (Sigma, 1:400) were used as secondary antibodies, followed by staining with 4,6-diamidino-2-phenylindole (DAPI; Sigma, 1:4000). Finally, the fluorescent signals were observed under Leica confocal fluorescence microscope (Leica, Germany) or Zeiss Axio Imager light microscope (Zeiss, Germany).
Cell viability assay
Joint capsule fibroblasts were seeded into 96-well plates at a density of 1 × 104 cells/well and treated with 0–10 μM PGE2 (Sigma, MO, USA) for 24 h. Then, the cells incubated with Cell Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan) for an additional 1 h. A microplate reader was used to detect absorbance at 450 nm to quantitatively measure cell viability. Assays were performed in triplicate.
Enzyme-linked immunosorbent assay (ELISA)
Cell samples or tissue homogenate were collected and centrifuged. The concentrations of key inflammatory cytokines including PGE2 (ARBOR ASSAYS, Michigan,USA), IL-1β, TNF-α, and IL-6 (MULTI SCIENCES, hangzhou, China) were quantified in the supernatants using specific ELISA kits following the protocols provided by the manufacturers. A microplate reader (Biotek Synergy2) equipped for 96-well plates was used to measure the absorbance at 450 nm.
EdU analysis
Joint capsule fibroblasts were plated into 96-well plates at a density of 1 × 104 cells/well and incubated with 0–5 μM PGE2 for 24 h. Then, the cells incubated with EdU for an additional 2 h. To assess cell proliferation, cells were processed using the Cell-Light EdU DNA Cell Proliferation Kit (RiboBio) according to instructions and analyzed using a fluorescence microscope (Leica) based on the images of randomly selected fields.
Transwell assay
Joint capsule fibroblasts were pre-treated with mitomycin C and then seeded at a density of 2 × 104 cells/well to the Transwell upper chambers (Costar; 8 μm pore size, 6.5 mm diameter). The appropriate lower chambers of the 24-well plate were filled with 600 μl medium containing 0–5 μM PGE2. Following a 24-h incubation period to allow for cell migration, non-migrated cells were gently removed from the membrane’s upper surface. The cells that migrated to the lower surface were then fixed in 4% paraformaldehyde, stained with 0.2% crystal violet (Sigma), and subsequently visualized and quantified under a Leica inverted microscope by counting the stained cells across several randomly selected microscopic fields.
Statistical analysis
Data analysis was performed using SPSS 22.0 statistical software (SPSS Inc., Chicago, IL, USA), with all results presented as mean ± standard deviation. Parametric data were first analyzed using the one-way analysis of variance (ANOVA) or Student's t-test, followed by Tukey's post hoc test for pairwise comparisons between groups. GraphPad Prism software 4.0 (GraphPad, CA, USA) was uesd for calculations. Differences were considered statistically significant when P < 0.05.


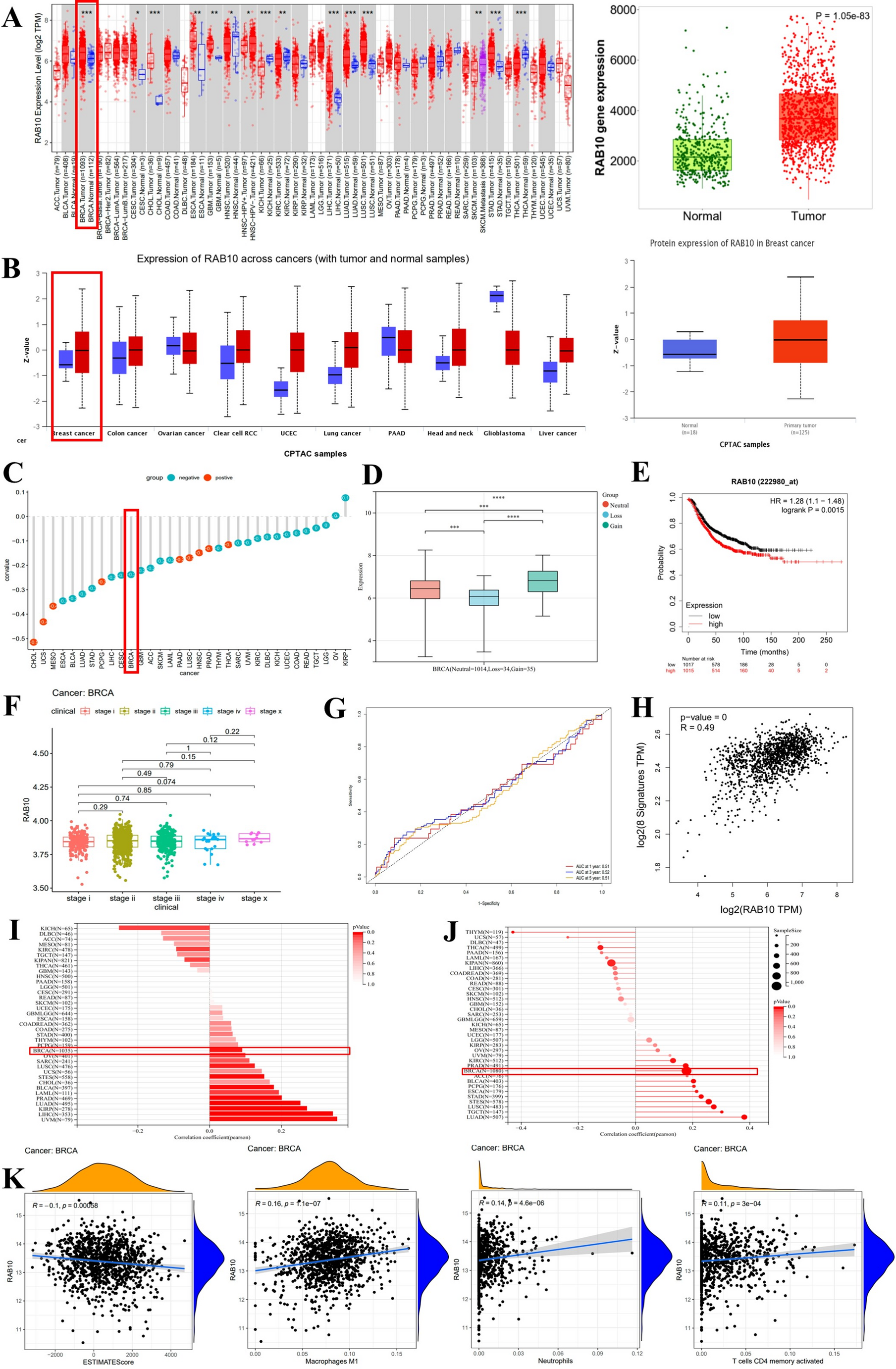
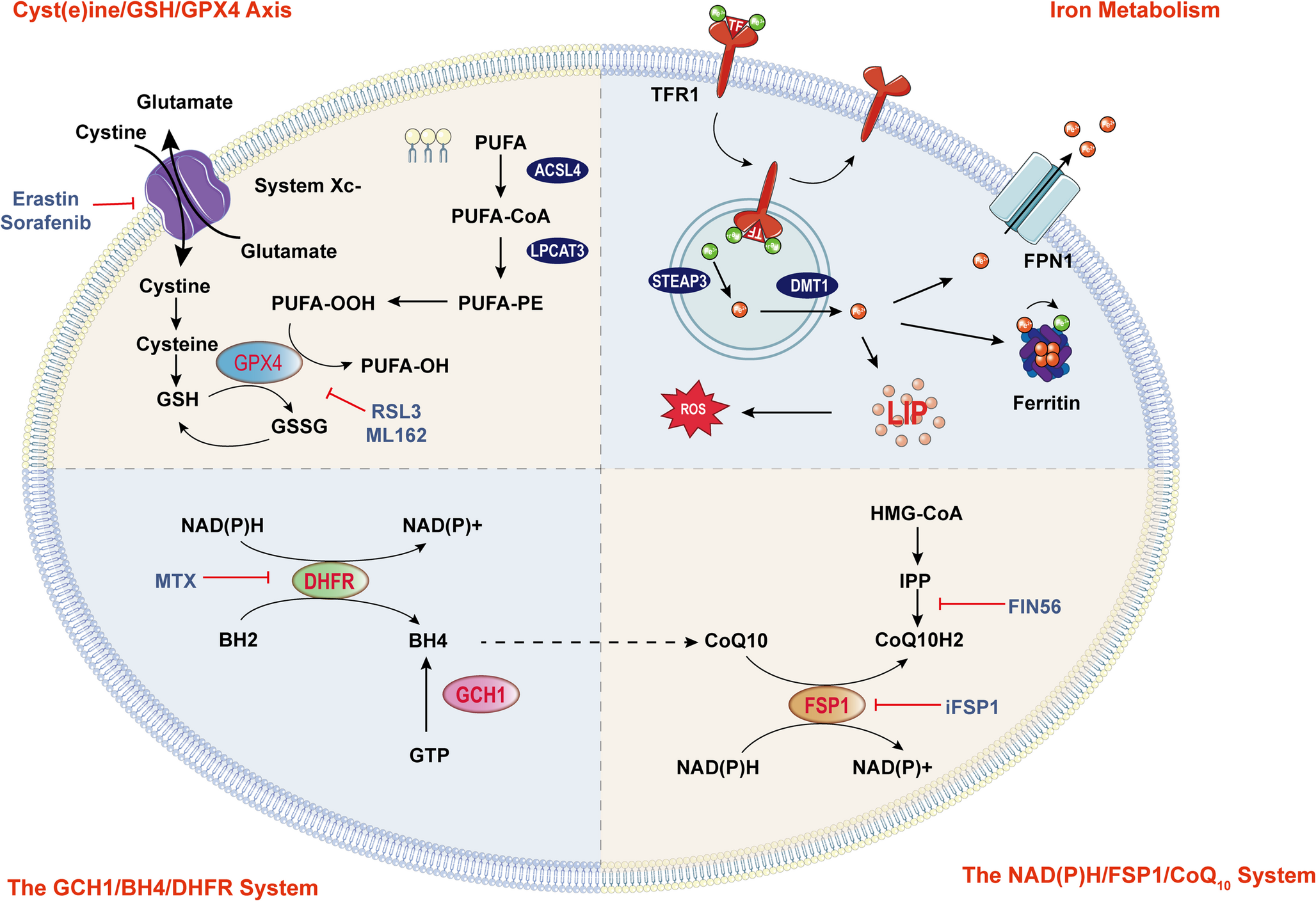
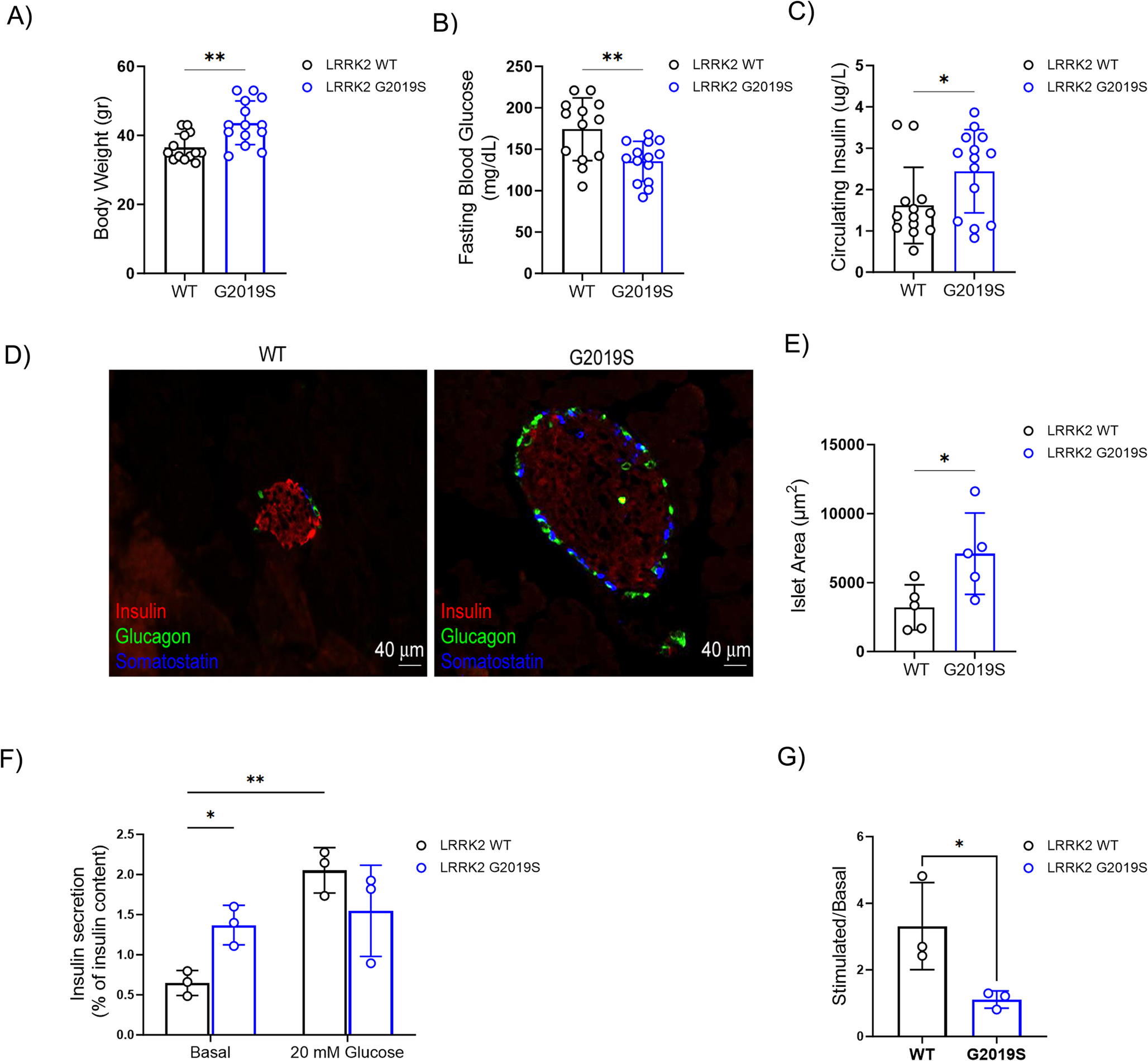
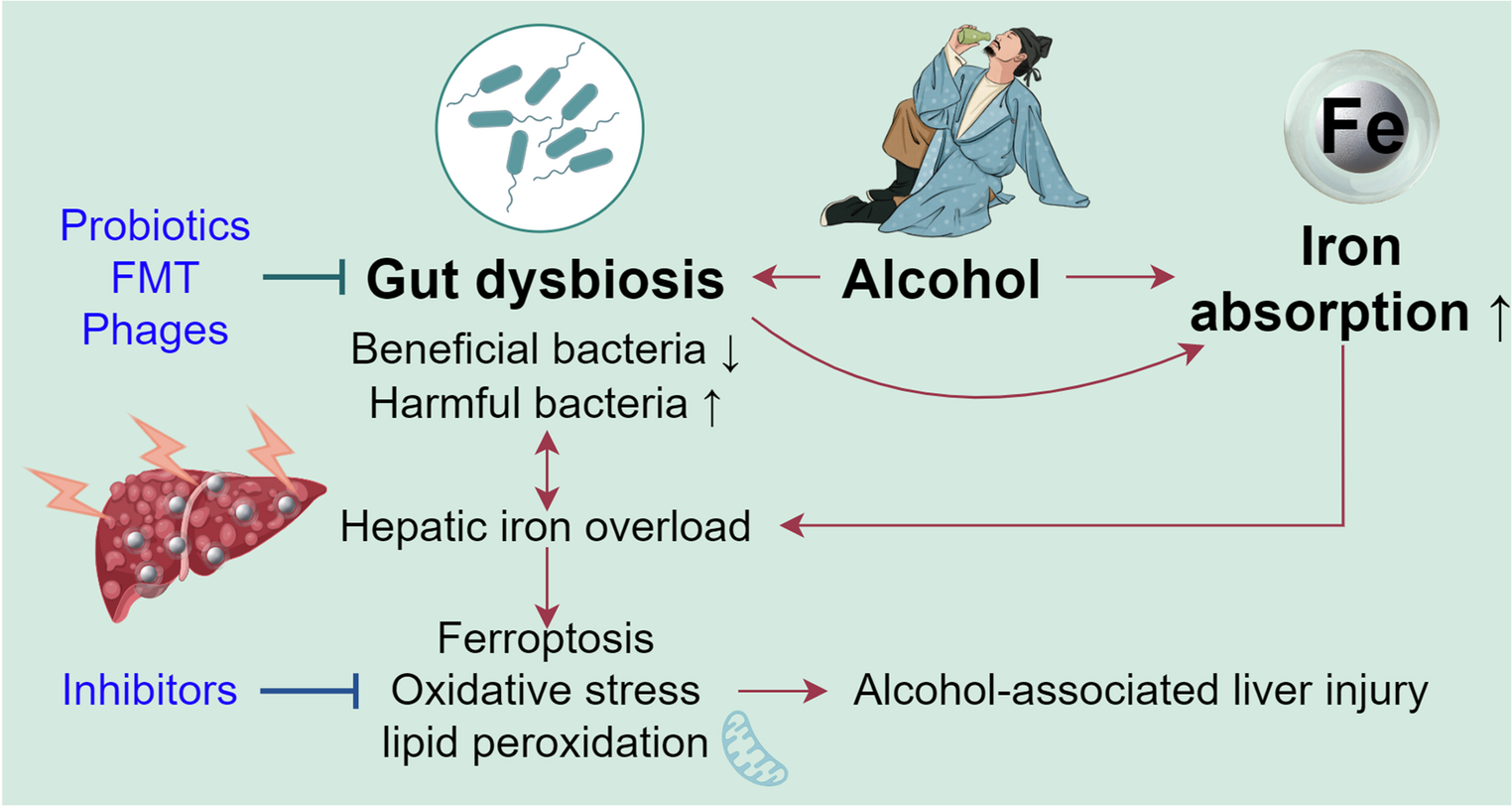
Comments (0)