Animal experiments
All animal procedures were conducted in accordance with institutional guidelines and approved by the Sun Yat-sen University Institutional Animal Care and Use Committee under protocol number SYSU-IACUC-2022-000887. Male mice aged 12–16 weeks were used in all experiments. This age range was selected to model the adolescent-to-young adult transition in humans, during which testicular torsion—a leading cause of ischemia-reperfusion injury (IRI)—is most prevalent. Its use also aligns with previous preclinical studies on testicular IRI and immune modulation [30, 31], enabling cross-study comparisons and enhancing translational relevance.
Wild-type (WT) C57BL/6 mice were purchased from Guangdong Medical Laboratory Animal Center. Dbh-Cre (MMRRC, 032081-UCD) mice were used for the chemogenetic and optogenetic experiments. All mice were maintained under controlled temperature (22 °C ± 1 °C with 30–70% humidity) and photoperiod (12 h light/dark cycle) in groups of 3–5 animals and were given free access to food and water.
All surgical procedures were conducted under isoflurane anesthesia (1–2% in oxygen) to minimize discomfort. Postoperatively, mice received one intraperitoneal injection of buprenorphine (0.3 mg in 100 µl saline) to manage pain. Animals were closely monitored for signs of distress, reduced mobility, or abnormal grooming behavior twice daily for three days post-surgery. Supportive care, including thermal support and hydration, was provided as needed. Humane endpoints were strictly observed, and any animal exhibiting prolonged distress or complications was euthanized in accordance with IACUC guidelines.
Testicular bilateral ischemia operation
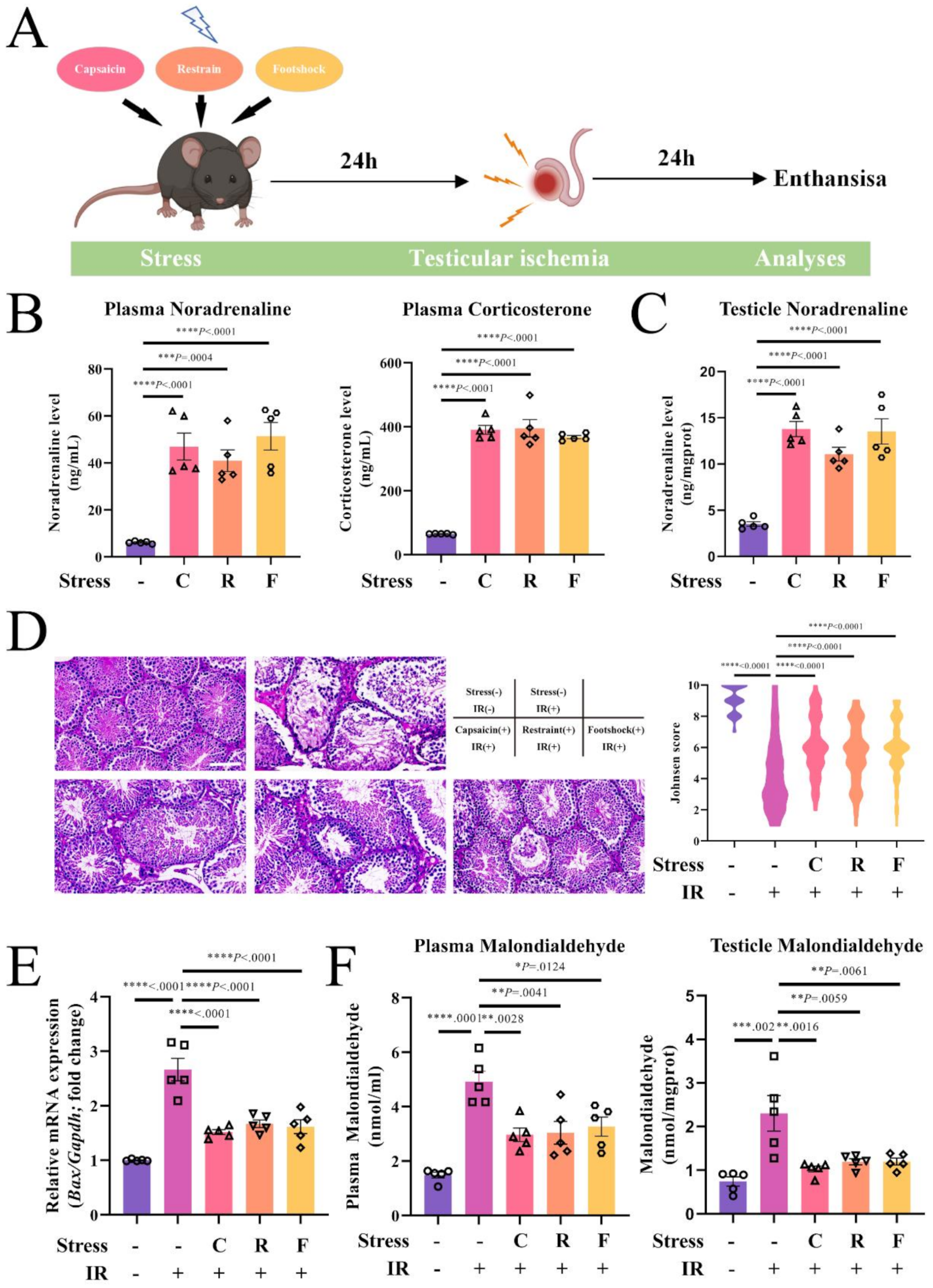
Mice were subjected to 60-min testicular bilateral ischemia followed by 24 h of reperfusion. Body temperature was maintained at 37.0 ± 0.5 °C with a servo-controlled temperature pad. The wound was sutured after visual observation of blood flow restoration. Sham-operated mice underwent the same procedure without clamping of the testicular pedicles. Twenty-four hours after testicular IR, the animals were euthanized, and blood plasma was collected by cardiac puncture in 2 ml Eppendorf tubes containing EDTA for malondialdehyde measurement. Next, we harvested the testes for histology and Bax mRNA measurement.
Stress model protocols
For the stress studies, the mice were exposed to stress on the first day and received testicular bilateral ischemia surgery 24 h later as described above. Restraint stress primarily engages psychological stress pathways with minimal sensory input, modeling emotion-driven SNS activation [32]. Foot shock introduces a mild nociceptive component, representing low-intensity physical stress [33,34,35]. In contrast, capsaicin induces robust nociceptive stimulation via TRPV1 activation, eliciting a high-intensity physiological stress response [36]. This gradation allowed us to assess the relationship between stressor intensity, SNS activation, and the extent of testicular protection. For restraint stress as previously described [37, 38], the mice were restricted in a ventilated 50 ml Falcon tube (Corning, #352070), in which they could rotate from a prone to supine position and back again but not turn head to tail, for 10 min. For electric foot shock stress as previously described [37, 39], the mice experienced a 10 min foot shock (0.5 mA for 2 s with a 60 s interval). For capsaicin stress as previously described [37, 40], mice were stimulated by subcutaneous capsaicin (Sigma, St. Louis, MO, USA) injection (10 mg/20 mL) in the dorsal aspect of the hind paw unilaterally. Twenty-four hours after IR, the animals were euthanized, and the blood plasma and testes were harvested as described in the previous section.
Viral vectors
rAAV2/9-hSyn-DIO-hM4D(Gi)-mCherry-WPRE-pA (Cat. No. S0193-9)
rAAV2/9-hSyn-DIO-mCherry-WPRE-pA (Cat. No. S1138-9)
rAAV2/9-hEf1a-DIO-hChR2(H134R)-mCherry-WPRE-pA (Cat. No. S0170-9)
rAAV2/9-hEf1a-DIO-mCherry-WPRE-pA (Cat. No. S0197-9)
rAAV2/2Retro Plus-hSyn-DIO-hM3D(Gq)-eGFP-WPRE-pA (Cat. No. S0260-2RP)
rAAV2/2Retro-hSyn-DIO-hM4Di(Gi)-eGFP-WPRE-pA (Cat. No. S0286-2RP)
rAAV2/2Retro Plus-hSyn-DIO-eGFP-WPRE-pA (Cat. No. S0276-2RP)
All viral vectors were obtained from Taitool Bioscience Co., Ltd., aliquoted, and stored at − 80 °C prior to use. Injections were performed at a final titer of 2.0 × 10¹² vg·ml⁻¹ for both brain and testes delivery.
Adrenalectomy
C57BL/6 mice were anesthetized, and small incisions were made on the back skin directly above each adrenal gland. Both adrenal glands were removed with a pair of curved forceps. Sham mice underwent the same procedures as the adrenalectomized (ADX) mice, except their adrenal glands were not removed. The body temperature of all experimental mice was maintained at 37.0 ± 0.5 °C during the surgery and recovery using a servo-controlled temperature pad. Postsurgery, 0.9% saline solution was provided instead of drinking water for both ADX and sham mice. All animals were permitted to recover for 7 days before testicular IR, during which time they were housed individually and handled. Any animals showing signs of illness or infection were removed from the study.
To ensure the integrity of adrenal function in sham-operated animals, we implemented the following exclusion criteria: animals exhibiting visible adrenal hemorrhage, basal serum corticosterone levels below 50 ng/ml [41, 42], or > 10% body weight loss within 72 h post-surgery were excluded from analysis.
Stereotactic surgery
Normal surgical procedures were followed for vector delivery and optic fiber implantation. In brief, mice were anesthetized with 3.0% isoflurane at the beginning of surgery and 0.5-1.0% isoflurane for the following surgical protocol. Mice were placed in a stereotaxic frame and maintained at 35 °C by a heating pad throughout the surgery. The following stereotaxic coordinates were used for virus and optic fiber delivery: for the RVLM: -6.96 mm from bregma, 1.30 mm from midline and 5.75 mm vertical from skull surface; for the LC: -5.40 mm from bregma, 0.80 mm from midline and 3.75 mm vertical from skull surface; for the NTS: -0.90 mm from bregma, 0.70 mm from midline and 4.40 mm vertical from skull surface.
For virus injection, a bilateral skull hole (1–2 mm2) was made for each mouse, through which a glass micropipette with a 5 μm tip diameter containing virus solution was lowered down to the targeted brain region. Approximately 300 nl of virus was injected at a speed of 50 nl per minute, which was controlled by a syringe pump (KD Scientific, Model 130). After virus injection, the micropipette was held at the target site for at least 15 min and then removed slowly. For optogenetics experiments, the optic fibers (diameter, 200 μm; NA, 0.37) were bilaterally implanted into the RVLM.
In vivo optogenetics and chemogenetics
For chemogenetic inhibition of catecholamine (CA) neurons in the LC, RVLM or NTS, we bilaterally expressed the hM4Di or EGFP (as the control) into these neurons in Dbh-Cre mice with the respective viruses. 14 days after virus injection, all mice were treated with the hM4Di agonist CNO (1 mg per kg of body weight, subcutaneous injection, Sigma‒Aldrich) 30 min before stress challenge and the testicular bilateral ischemia surgery was performed 24 h later.
To activate the CA neurons of the RVLM, ChR2 or mCherry was selectively expressed in these neurons, and optic fibers were delivered into the RVLM in Dbh-Cre mice for light stimulation. 14 days after virus injection, laser stimulation (5-ms pulses, 10 Hz, 5 mW (measured at the tip of optic fibers)) was performed for 10 min, and testicular bilateral ischemia surgery was performed 24 h later.
To determine whether activation of direct SNS input to testes could drive the protective effect, the retrograde hM3Dq or EGFP was delivered bilaterally into the testes in the Dbh-Cre mice. 21 days after the virus injection, all the mice are treated with CNO (1 mg per kg of body weight, subcutaneous injection, Sigma‒Aldrich) and the testicular bilateral ischemia surgery was performed 24 h later.
To silence the SNS input to testes, the retrograde hM4Di or EGFP was delivered bilaterally into the testes in the Dbh-Cre mice. 21 days after the virus injection, all the mice are treated with CNO (1 mg per kg of body weight, subcutaneous injection, Sigma‒Aldrich) before stress challenge and the testicular bilateral ischemia surgery was performed 24 h later.
Testicular macrophage depletion
In testes-resident macrophage depletion experiments, clodronate liposomes (LIPOSOMA research, Haarlem, the Netherlands) were injected locally into both testes of mice at a dose of 10 µl (5 mg/ml). This dose was chosen based on our own pilot data and earlier protocols. We injected the same volume of control liposomes (PBS) as a control. Clodronate, an effective macrophage scavenger, was incorporated into liposomes as previously described [43]. Clodronate liposomes have been used extensively to specifically deplete macrophages in vivo by inducing apoptosis in these cells without affecting other cell types. To determine the effect of testes-resident macrophage depletion, 7 days after administration, mice were sacrificed, and frozen testes were sectioned and labeled with anti-mouse F4/80 antibodies against macrophages. After 7 days of administration, the animals were subjected to testicular IRI as described previously.
Interstitial cell collection and flow cytometry
Mice were killed by cervical dislocation under deep isoflurane anesthesia, and the testes were retrieved, minced, and enzymatically digested into single-cell suspensions. Briefly, the decapsulated testes were incubated with 50 µg/mL DNase I (Roche Diagnostics, cat. 10104159001) and 1 mg/ml type IV collagenase (Gibco, cat. 17104-019) in DMEM in a shaking water bath at 37 °C for 45 min. The enzymes were inactivated by adding ice-cold PBS, and the tubule fragments were allowed to settle for 4 min. Then, the supernatant containing predominantly interstitial cells was collected and filtered through a 35-µm cell strainer cap to remove the seminiferous tubules to obtain a single cell suspension and centrifuged at 750 × g for 5 min at 4 °C, and the resulting pellet was resuspended in FACS buffer (5 mM EDTA, 0.02% sodium azide and 1% fetal bovine serum in PBS). Red blood cell lysis buffer was added to the cell pellet; after disrupting the pellet, the cells were incubated for 5 min at room temperature, and cold PBS was added to stop the reaction. Following erythrocyte lysis, cells were resuspended in FACS buffer. Nonspecific binding to low-affinity Fc receptors was blocked by incubating the cells (106) with unconjugated CD16/32 antibody (BioLegend, cat. 156603) with FcR blocking buffer (2% rat serum in FACS buffer) for 10 min at 4 °C. After blocking, the cells were subsequently incubated with antibody cocktails for 30 min at 4 °C and then washed with FACS staining buffer. The following antibodies were used: Alexa Flour 700-conjugated anti-CD45 (BioLegend, cat. 103128), eFluor 450-conjugated anti-F4/80 (eBioscience, cat. 48-4801-82), PE/Cy7-conjugated anti-CD11b (BioLegend, cat. 101216), APC-conjugated anti-Ly6C (BD Pharmingen, cat. 560595), PE/Dazzle 594-conjugated anti-CD11c (BioLegend, cat. 117348), PE/Cy5-conjugated anti-MHC class II [IA] (BioLegend, cat. 107612), PE-conjugated CD206 (BioLegend, cat. 141705), APC/Cy7-conjugated anti-CD86 (BioLegend, cat. 105029). Cells were stained with Fixable Viability Dye eFluor 506 (eBioscience, cat. 65-0866-14) to label the dead cells. Samples were analyzed with CytoFLEX S (Beckman, Indianapolis, IN, USA) flow cytometers and CytExpert (Beckman, Indianapolis, IN, USA) software.
Animal tissue harvesting and plasma analysis
Mice were euthanized, and blood was harvested by cardiac puncture. Organs were harvested and fixed with 4% paraformaldehyde (PFA). Blood hematology was performed on blood collected in EDTA. Plasma analysis was performed using commercial kits according to the manufacturer’s instructions for malondialdehyde (Jiancheng Bioengineering, Cat. A003-1-2), corticosterone (Catalog No. 3782, MEIMIAN) and norepinephrine (Catalog No. 11699, MEIMIAN).
Quantitative real-time PCR
Total RNA was extracted from testes and testicular interstitial cells using a combination of TRIzol™ reagent (Invitrogen) and the Qiagen RNeasy Mini system (Qiagen, UK) according to both manufacturers’ instructions, and RNA concentration was determined based on spectrophometric determination of the 260/280 ratio using a NanoDrop 1000 (Thermo Fisher Scientific, Wilmington, DE, USA). cDNA was generated from the resultant tissue RNA using the High-Capacity RNA-to-cDNATM Kit (Thermo Fisher Scientific) as described by the manufacturer. The resultant cDNA was then subjected to quantitative real-time PCR (qPCR) to determine the relative mRNA expression of Bax and Gapdh using LightCycler 480 SYBR Green I Master Mix (Roche Diagnostics GmbH, Mannheim, Germany) on a Light Cycler 480 Detection System (Roche Diagnostics GmbH, Mannheim, Germany). The primers used for gene assessment were as follows: Bax (fwd)– AGA CAG GGG CCT TTT TGC TAC, (rev)– AAT TCG CCG GAG ACA CTC G; Gapdh (fwd)– AGG TCG GTG TGA ACG GAT TTG, (rev)– GGG GTC GTT GAT GGC AAC A.
Plasma corticosterone and norepinephrine measurement
To investigate whether the classic stress responses are activated by stress stimulation, three approaches to model stress in C57BL/6 mice are described above. 60 min later, the mice were quickly anesthetized with isoflurane, and blood was immediately collected into EDTA-coated tubes from the orbital sinus. Plasma was then prepared by centrifuging heparinized blood at 7,000 × g for 5 min at 4 °C and stored at − 80 °C until analysis. All of these experiments were conducted between 1 and 5 PM. Baseline blood was withdrawn under short isoflurane anesthesia, and blood was immediately collected from the orbital sinus. For adrenalectomy studies, the adrenalectomized mice underwent the same procedures as above 7 days after the surgical procedure. Commercial ELISA kits were used to measure plasma corticosterone (Catalog No. 3782, MEIMIAN) and norepinephrine (Catalog No. 11699, MEIMIAN) levels in a blinded fashion. Concentrations of plasma corticosterone and norepinephrine were measured by ELISA kits following the manufacturer’s instructions. Finally, the absorbance of each well was determined by a microtiter plate reader (Thermo Scientific, Vantaa, Finland).
Norepinephrine measurement in the testes
We examined whether stress treatments or short trains of light pulses at 10 Hz for 10 min could activate the testicular sympathetic nervous system in Dbh-ChR2 mice. For optogenetic stimulation, we briefly anesthetized the Dbh-ChR2 mice with isoflurane and attached them to the fiber optic patch cable. After a short recovery from isoflurane, mice were exposed to light stimulation as described above. Then, they were put back in their original cages and euthanized 60 min later. Immediately after decapitation, the testes were removed from the animal, frozen in liquid nitrogen and stored at − 80 °C until analysis. We homogenized the testes samples in ice-cold PBS with protease inhibitor cocktail (Sigma) and centrifuged them at 14,000 × g for 10 min at 4 °C to remove insoluble components. Values were normalized using total protein levels measured with the Pierce BCA Protein Assay (Thermo Fisher Scientific, Rockford, USA, 23225) following the manufacturer’s instructions. Tissue norepinephrine levels were measured using ELISA, as described above. Dbh-ChR2 mice attached to the fiber optic patch cable but without light stimulation served as controls. The chemogenetic activation was the same as above. Finally, the absorbance of each well was determined by a microplate reader (Thermo Scientific, Vantaa, Finland).
Determination of malondialdehyde production
Blood was obtained by cardiac puncture and collected in a separator EDTA-coated tube. Plasma was prepared by centrifuging heparinized blood at 7,000 × g for 5 min at 4 °C. The plasma malondialdehyde contents were evaluated spectrophotometrically by using malondialdehyde detection kits (Jiancheng Bioengineering, Cat. A003-1-2), measuring the presence of thiobarbituric acid reactive substances (TBARS) [44]. The absorbance was determined by a spectrophotometer (Thermo Scientific, Vantaa, Finland) at 532 and 520 nm. The amount of lipid peroxides was calculated as TBARS of lipid peroxidation.
Histological analysis of mouse testes
Following completion of the in vivo experiments, mice were euthanized and perfused transcardially with 50 mL of heparinized saline followed by 100 mL of freshly prepared 4% paraformaldehyde in 100 mL of sodium phosphate buffer (pH 7.4). Harvested testes were preserved in Bouin’s solution (7.5 mL saturated picric acid, 2.65 mL glacial acetic acid and 2.5 mL 7% formaldehyde), hydrated, postfixed in 70% alcohol, and embedded in paraffin blocks. Tissue Sect. (4 μm) from paraffin-embedded mouse testes samples were subjected to routine hematoxylin and eosin (H&E) staining. Hematoxylin and eosin (H&E) staining was performed according to standard protocols. The testicular tissue was evaluated in random order with standard light microscopy by an investigator blinded to the treatment group of each sample. For the evaluation of overall testicular damage, we used the criteria formulated by Johnsen [45]. The Johnsen score, a ten-point scoring system, is based on the premise that with testicular damage, there is successive disappearance of the most mature cell type, with progressive degeneration of germinal epithelium, with the disappearance of spermatozoa and spermatids, then spermatocytes and finally Sertoli cells, in that order.
Microscopy and image processing
In the monosynaptic tracing experiments, major pelvic ganglia in reporter mice were confirmed with a fluorescent stereomicroscope (Leica M205FA, Leica Biosystems Inc., Buffalo Grove, IL). Imaging was performed with a zoom factor of 1.6×. Images of tyrosine hydroxylase labeling were collected for all samples. Images from the GFP fluorescent channel were pseudocolored green.
For DREADD tracing studies, neurons labeled with either EGFP in major pelvic ganglia in Dbh-Cre mice (received intratesticular injections) were imaged manually by examining on an epifluorescence microscope (Zeiss). Every 25 μm major pelvic ganglia section was collected. Individual tissue sections were scanned with a 10× objective using Zeiss Axio Scan Z1 slide scanner or were directly examined with Zeiss LSM 880/980 microscope. Lastly, DAPI and GFP channels were turned on and primary sympathetic innervations in the testes originate from the major pelvic ganglion were imaged for every sections.
Tissue handling for frozen sections
To determine the neural activity under different stresses, the brain was collected 60 min after stimulation. Similarly, for verification of hM4Di in the regulation of neural activity, the mice received CNO treatment and experienced capsaicin stimulation 30 min later. Then, brain tissue was collected 60 min after stress stimulation for immunofluorescence staining. For verification of ChR2 stimulation of neurons, brain tissue was collected 60 min after 10 Hz blue light stimulation for 10 min. For retrograde labeling of the innervating neuronal populations, three weeks after AAV injection, the ganglia and spinal cord were harvested from the mice.
Mice were euthanized and perfused intracardially with cold phosphate-buffered saline (PBS), followed by 4% paraformaldehyde (PFA) in PBS with 0.2% picric acid. Harvested testes were immersed in 4% PFA for 3 h at room temperature. Fixed testes were cryoprotected by successive overnight incubations at 4 °C in 15% and 30% sucrose solutions. For immunostaining using frozen sections, samples were embedded in Tissue-Tek optimum cutting temperature (O.C.T.) compound (Sakura Finetek, USA) and snap-frozen in isopentane precooled in liquid nitrogen. The samples were cryosectioned to a thickness of 25 μm at −20 °C on a freezing cryostat (Leica CR 1950) and mounted on glass slides. All frozen brain sections in this study were cut at a uniform thickness of 25 μm using a cryostat, which we found optimal for maintaining structural integrity and signal resolution during histochemical analyses [46,47,48]. The sections of tissues were used for immunofluorescence staining and fluorescence microscopy.
Immunofluorescence staining
All procedures were performed at room temperature with 5 min washes unless otherwise specified. For immunofluorescence staining, the slides were then blocked in blocking buffer (0.2% Triton X-100, 0.05% sodium azide, 4% bovine serum albumin in PBS) for 1 h at room temperature, incubated with primary antibodies overnight at 4 °C, and incubated with secondary antibodies for 2–4 h at room temperature. Brain sections were incubated with the following primary antibodies: rabbit anti-dopamine beta hydroxylase antibody (Abcam, ab209487, 1:1500), mouse anti-c-Fos antibody (Abcam, ab208942, 1:1000), and rabbit anti-mCherry antibody (CST, 43590, 1:100). Ganglion sections were incubated with primary antibody: rabbit anti-GFP (1:1000, cat. AB152, Santa Cruz). Testes sections were incubated with the following primary antibodies: rat anti-F4/80 (general macrophage marker; Abcam, ab15580, 1:100), rabbit anti-beta 2 adrenergic receptor (Abcam, ab182136, 1:100) and rabbit anti-tyrosine hydroxylase (CST, AB152, 1:100). The following secondary antibodies were used: Alexa Fluor 488-donkey anti-mouse IgG (1:500, Jackson ImmunoResearch Laboratories) and Alexa Fluor 594-goat anti-rabbit IgG (1:500, Jackson ImmunoResearch Laboratories). Fluorescence images were acquired using a confocal microscope or an automated slider scanner and processed using ImageJ (NIH). The locations of labeled neurons and outlines of the brain regions were manually defined according to the mouse brain atlas [49]. Cell numbers were counted automatically using commercial Imaris software (Bitplane). 4′,6-Diamino-2-phenylindole (DAPI; Roche) was used as a counterstain for the nucleus.
TH fiber proximity analysis
Confocal imaging was performed on a Zeiss LSM 980 system using a Plan-Apochromat 40×/1.3 Oil objective, acquiring 15-layer z-stacks (1 μm step size) across 3 × 3 tiled fields per section at 1024 × 1024 resolution (0.22 μm/pixel) with 405/488/561 nm laser lines. For 3D quantification, maximum intensity projections were generated in ZEN Blue 3.5 followed by channel separation.
Microscopy and c-Fos quantification
In the monosynaptic tracing experiments, a Leica M205FA (Leica Biosystems Inc., Buffalo Grove, IL) was used to image all relevant structures. Images were captured using Carl Zeiss LSM 800/880 confocal microscopy and then processed by ZEN software. c-Fos is a well-established immediate-early gene and is widely used as a surrogate marker of neuronal activation in response to acute stimuli, including stress [50]. c-Fos immunoreactivity was used to determine the effects of stress, chemogenetic, and optogenetic, on neuronal activity. Sixty minutes after the appropriate stimulation, mice were sacrificed and perfused as described above. c-Fos expression was analyzed and quantified as follows: Coronal sections at ∼160 μm intervals were photographed at 10× magnification and montaged with ImageJ to preserve anatomical landmarks. c-Fos+ neurons in each section separated by color channel, and artifacts were removed. An area tool was used to select the biopsy region and counted. Expressed as the cumulative sum of c-Fos+ neurons within the relevant regions for each animal.
Illustrations
The graphical abstract was created with BioRender.com. All figures were created using Illustrator software (Adobe).
Statistical analysis
All statistical analyses were performed using GraphPad Prism 9. Comparisons between two groups were evaluated using unpaired two-tailed Student’s t-tests, assuming equal variances unless otherwise noted. One-way or two-way ANOVA followed by Tukey’s post hoc test was used for multiple group comparisons involving a single independent variable. Data are presented as mean ± SEM unless otherwise indicated. Sample sizes (n), exact P-values, and test types used for each comparison are detailed in the figure legends.



Comments (0)