Establishment of I/R models and drug administration
All animal-related protocols in the present study were approved by the Animal Ethics Committee of Southeast University (Nanjing, China, 20240219008) and conducted in accordance with the National Institutes of Health (NIH) guidelines. C57BL/6 or ICR mice (8 weeks old, male) were used for establishing the I/R models. These mice were housed in pathogen-free conditions with constant temperature of 20 ± 2 °C, humidity of 50–70%, and a regular 12 h light–dark cycle. Besides, standard chow and water were timely applied for these animals.
After 1 week of acclimatization, these mice received intraperitoneal anesthesia with sodium pentobarbital and then randomly underwent the indexed I/R surgery as previously described (n = 12) [21] or sham operations (n = 12). Briefly, a 7–0 surgical suture was used for ligating the left anterior descending (LAD) coronary artery with a slipknot, one thread end of which was exposed outside the thoracic cavity. One hour later, the slipknot was released to allow reperfusion of the heart. Conversely, sham-operated mice underwent all the surgical procedures but without ligation of the proximal LAD coronary artery. Notably, these mice were maintained on a thermal plate with a constant temperature of 37 °C during the whole procedure.
To better evaluate the effectiveness and safety of XTS, we dissolved this compound (100 mg/kg, HY-W011527, Med Chem Express, China) in dimethyl sulfoxide (DMSO, HY-Y0320, Med Chem Express, China), which was then diluted with sterile saline to ensure a final DMSO concentration of 5%. The prepared solution was administered to the mice via intraperitoneal injection 1 h prior to reperfusion, and then continued once daily (labeled Sham + XTS or I/R + XTS). For the control groups, an equal volume of saline solution containing 5% DMSO alone was administered similarly to the residual mice (denoted as Sham + DMSO or I/R + DMSO). Additionally, to provide a more comprehensive understanding of its effects, our animal experiments were conducted with these interventions at five different time points concurrently.
Echocardiography
Transthoracic M-mode and two-dimensional echocardiography was performed to assess the cardiac functions and left ventricular (LV) structures of these mice at 1 day, 7 days, and 14 days after surgery, respectively, using the Vevo 2100 Imaging System (FUJIFILM VisualSonics, Toronto, Canada) with a 30-MHz phased array linear transducer. All mice were anesthetized by inhalation of 3% isoflurane initially and maintained with 1.5% isoflurane during examination. The examined items mainly included the left ventricular internal diameter at end-diastole (LVIDd), internal diameter at systole (LVIDs), end-diastolic volume (EDV), and end-systolic volume (ESV). The LV ejection fraction (EF%) and fractional shortening (FS%) were then calculated as described previously [22].
2,3,5-Triphenyltetrazolium chloride (TTC)–Evans blue double staining
TTC–Evans blue staining was used for identifying the ischemic myocardium and calculating the infarct size. Twenty-four hours after surgery, the mice were euthanized and injected with 1.5% Evans blue dye (approximately 0.2 mL for each 25 g mouse) through the thoracic aorta after the occlusion of the LAD artery. These hearts were then harvested rapidly and rinsed with phosphate-buffered saline (PBS) to remove any superficial dye. Of note, the tissues were frozen at −20 °C for 30 min, followed by uniform slicing from the ligation point to the apex, with thickness of 1 mm. Subsequently, these tissue sections were incubated with 1% TTC solution for staining, then observed under a stereomicroscope for image acquisition. The results were quantitatively analyzed by using ImageJ software.
Histology and immunofluorescence staining
The infarcted heart tissues were fixed with 4% paraformaldehyde and then were sectioned into 3-μm-thick slices after embedding with paraffin. These slices underwent the following staining:
(1)
For cardiomyocyte proliferation measurements, the sections were incubated with the following primary antibodies: cTnT (2 ug/ml, ab8295, Abcam, UK) and Ki67 (1:100, ab15580, Abcam, UK) after blocking with 5% bovine serum albumin (BSA) containing 0.2% Triton X-100. Thereafter, these sections were then incubated with species-matched Alexa Fluor 488 or Alexa Fluor 594 secondary antibodies (1:500, Biosharp, China) and counterstained with 4′,6-diamidino-2-phenylindole (DAPI; G1012-10ML, Servicebio Technology, China). Finally, the stained tissue sections were subsequently observed using Case Viewer (Wuhan Servicebio Technology Co., LTD).
(2)
Wheat germ agglutinin (WGA; G1730-100UL, Servicebio Technology, China) and DAPI (G1012-10ML, Servicebio Technology, China) staining was used to determine cross-sectional area (CSA). The stained tissue sections were subsequently observed using Case Viewer (Wuhan Servicebio Technology Co., LTD). Cardiomyocyte CSA was analyzed by using ImageJ software (NIH, Bethesda, MD, USA; http://rsb.info.nih.gov/nih-image/).
(3)
Cardiomyocyte apoptosis was evaluated using a terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining kit (G1505-50 T, Servicebio Technology, Wuhan, China) following the manufacturer’s guidelines. The stained tissue sections were subsequently observed using Case Viewer (Wuhan Servicebio Technology Co., LTD).
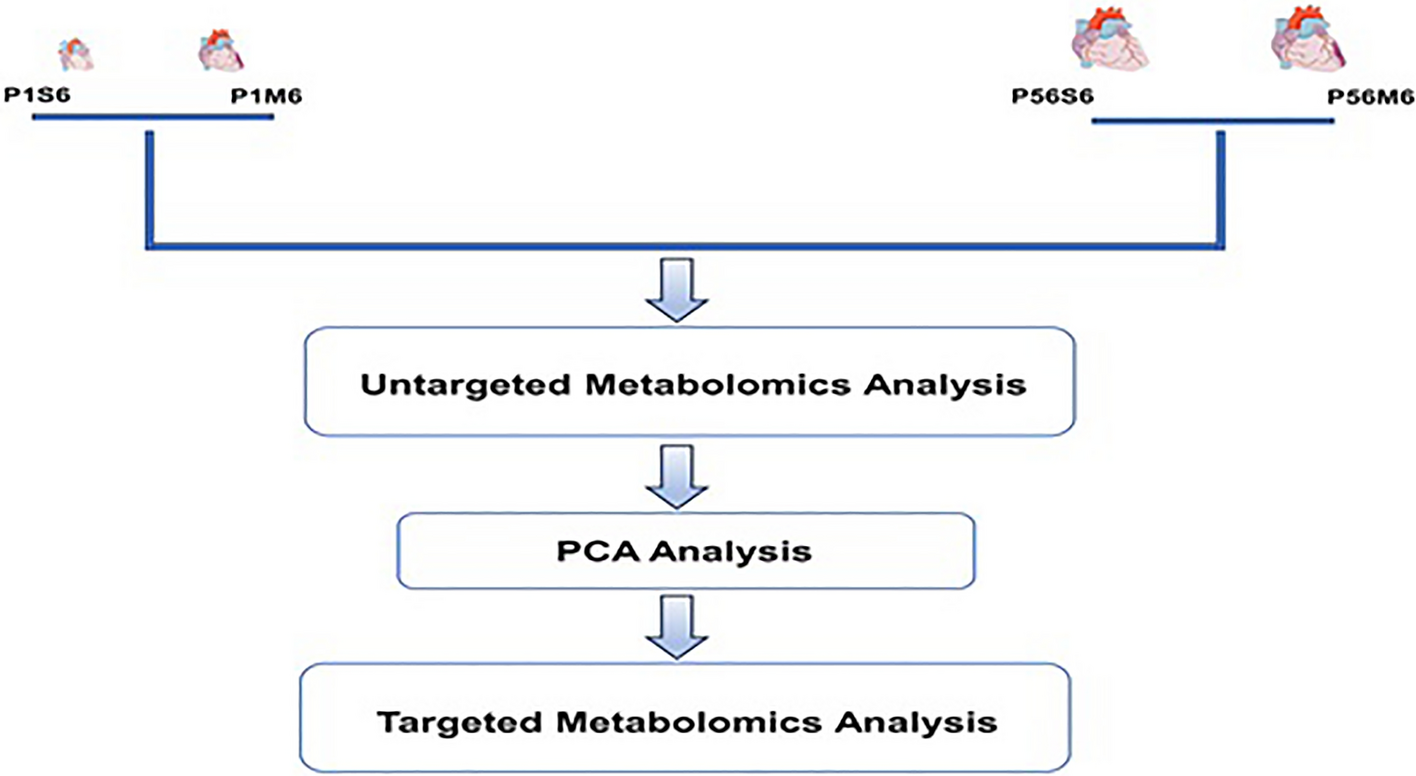
Untargeted metabolomics analysisSample preparation
Cardiac tissue samples were thawed on ice. A 30-mg portion of the tissue was then homogenized with 400 μL of extraction solution (methanol:acetonitrile = 3:1, precooled to −40 °C). The mixture was vortexed for 5 min, subjected to ultrasonic treatment in an ice–water bath for 10 min, and incubated at 4 °C for 2 h. The supernatant after centrifugation (4 °C, 12,000 rpm for 15 min) was carefully collected and then vacuum concentrated to dryness. Moreover, the residue was reconstituted with 100 μL of 50% methanol aqueous solution (methanol:water = 1:1, v/v), vortexed for 3 min at 4 °C and 2000 rpm, and centrifuged again at 12,000 rpm and 4 °C for 15 min. The resulting supernatant was collected for mass spectrometry analysis.
LC–MS/MS analysis
Sample separation was carried out using an ultra-high performance liquid chromatography (UHPLC) system equipped with an HSS T3 chromatographic column at 40 °C (injection volume: 2 μL, flow rate: 0.3 mL/min). Each sample was analyzed in both positive and negative ion modes using electrospray ionization (ESI). After separation, the samples were analyzed by a Thermo QE HF-X mass spectrometer.
Data processing
Peak identification and extraction were performed using Compound Discoverer version 3.3. Missing values were imputed using linear regression, and the data were normalized based on the maximum median peak area across all samples. Statistical significance testing and classification analyses were conducted using R and Python programming languages. Differential metabolites were identified based on a fold change (FC) threshold of ≥ 2 and a P-value <0.05. The identified metabolites, categorized as upregulated or downregulated, were further analyzed through principal component analysis (PCA) and clustering to ensure a comprehensive understanding of the metabolic profiles.
Targeted metabolomics analysisSample preparation
Cardiac tissue samples were prepared similarly as above-mentioned.
LC–MS analysis
Multiple reaction monitoring (MRM) analysis was performed using a Thermo Vanquish UHPLC system coupled with a TSQ Altis high-resolution mass spectrometer (Thermo Fisher Scientific, USA). Data acquisition was controlled by XCalibur 4.4 software (Thermo Fisher Scientific), and data processing was performed using TraceFinder 5.1 software (Thermo Fisher Scientific). Ion channel information is provided in the Supplementary Table.
Isolation of neonatal rat cardiomyocytes (NRCMs), establishment of H/R model, and treatments
NRCMs were isolated and cultured as previously described [15]. Briefly, heart tissues from new-born Sprague–Dawley rats (1–2-day-old) were cut into fragments of 1 mm3 and incubated with Hank’s balanced salt solution (HBSS) containing 0.1% trypsin and 0.05% collagenase type II for further digestion. After filtration and 2 h of differential adhesion, the isolated NRCMs were cultivated in Dulbecco’s modified Eagle medium (DMEM) enriched with 10% fetal bovine serum (FBS) for 48 h and replaced with a serum-free medium for 12 h before treatments. For establishing the hypoxia/reoxygenation (H/R) model, these NRCMs were first incubated in FBS-free and glucose-free 1640 medium (#11966-025, Gibco, USA) under hypoxic conditions (94% nitrogen, 5% carbon dioxide, and 1% oxygen). After a 12-h hypoxic period, the NRCMs were transferred to DMEM supplemented with 10% FBS and then cultured in an incubator with 5% CO2 and 95% nitrogen for reoxygenation, which was maintained for 24 h. Before exposure to hypoxia and reoxygenation, the metabolites identified by the above-mentioned metabolomics analysis (XTS, 10 μM) were provided to these NRCMs in either the normoxia or H/R group for further exploration. Of note, XTS should be applied dissolved in DMSO. Besides, the parallel control group received an equal volume of empty DMSO to avoid confounders.
The H9c2 cell line, procured from the Shanghai Institute of Cell Biology at the Chinese Academy of Sciences (Shanghai, China), was cultivated under analogous conditions to those used for NRCMs. Upon achieving 70% confluence, the culture medium was swapped for a serum-free version, which was maintained for 12 h. Subsequently, the cells were subjected to a 6-h period of hypoxia, followed by a 4-h phase of reoxygenation. Prior to this hypoxic/reoxygenation process, varying concentrations of XTS or DMSO were administered as previously described.
Cell viability assay
Cell viability was assessed using the CCK-8 assay kit (BS350A, Biosharp, China) and Trypan Blue staining kit (ST798, Beyotime, China) according to the manufacturers’ instructions. After incubating for 1–4 h, the absorbance at 450 nm was measured for assessing the cell viability. Besides, these transfected NRCMs and H9c2 cells (approximately 1 × 105) were stained using Trypan Blue, then counted using a microscope (Nikon, Japan). Afterwards, the cell viability was determined according to the formula (number of viable cells/total number of cells) × 100%, providing a clear and direct measure of cell health and survival rate.
Measurement of drug affinity responsive target stability (DARTS)-LC/MS
DARTS-LC/MS was used to pinpoint potential endogenous targets of XTS in H9c2 cells as previously described [23]. Peptide separation and analysis were performed using an Easy-nLC 1200 system coupled to an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific). Raw MS data were processed with MaxQuant 1.5.4.1 (MS tolerance: 5 ppm; MS/MS tolerance: 20 ppm) and searched against the UniProt Database (2016_07 release, 70,630 entries) along with the MaxQuant Proteomics Contaminant Database for protein identification.
RNA-seq analysis
Following treatment with DMSO or XTS (10 μM), these H9c2 cells (1 × 106) which had undergone H/R injury were preserved in liquid nitrogen for subsequent RNA-Seq analysis, conducted by Beijing Novogene Biotechnology Co., Ltd. and Shanghai NewCore Biotechnology Co., Ltd., as previously described [24].
Cellular thermal shift assay (CETSA)
H9c2 cells were seeded in two 10-cm2 culture dishes. One dish was treated with 10 μM XTS, while the other received an equivalent volume of DMSO as a control. After incubation, cells were harvested and resuspended in PBS. The resuspended cell aliquots were divided into six microtubes and subjected to thermal challenge at seven temperature gradients ranging from 40 to 55 °C for 3 min. Subsequently, the samples were flash-frozen in liquid nitrogen. Post-heating, three freeze–thaw cycles were performed using liquid nitrogen to ensure complete cell lysis. The lysates were centrifuged at 12,000×g for 20 min at 4 °C to collect the supernatant, and equal volumes of proteins were analyzed by Western blotting using target-specific antibodies.
Detection of oxidative stress markers
The concentrations of relevant biomarkers with respect to oxidative stress in the NRCMs were measured using the corresponding commercial kit, including malondialdehyde (MDA, # A003-4-1), lactate dehydrogenase (LDH, # A020-2-2), and superoxide dismutase (SOD, # A001-3-2), which were all purchased from Nanjing Jiancheng Biotechnology Research Institute (China) and performed following the manufacturer’s instructions.
ATP detection
ATP levels were measured using an ATP Bioluminescence Kit (A22066, Invitrogen, USA,) following the manufacturer’s instructions. Heart tissues or the NRCMs were lysed first and added to a 96-well plate, respectively. The absorbance at 560 nm was measured, then ATP levels were calculated on the basis of a standard curve.
Measurement of ROS and mitochondrial ROS (mROS)
ROS and mROS levels were measured using the H2DCFDA probe (HY-D0940, MedChemExpress, China), MitoSOX™ Red mitochondrial superoxide indicator (HY-D1055, Med Chem Express, China) and dihydroethidium (DHE, HY-D0079, Med Chem Express, China) according to the manufacturer’s instructions, respectively. All images were acquired using an inverted fluorescence microscope (BZ-X810, KEYENCE, Japan) or a confocal microscope (FV3000, Olympus, Japan) and analyzed with ImageJ software.
Detection of mitochondrial membrane potential (Δψm)
The mitochondrial membrane potential (Δψm) of these NRCMs was assessed using the JC-1 fluorescent probe (C2005, Beyotime, China), adhering to the manufacturer’s guidelines. Under a fluorescence microscope, the cells were observed with the excitation wavelength set at 488 nm to reveal strong green fluorescence and at 594 nm to reveal striking red fluorescence.
Measurement of the mitochondrial oxygen consumption rate (OCR)
The mitochondrial oxygen consumption (OCR) of NRCMs was evaluated using a MitoStress Test kit (cat. no. 103015-100; Agilent) (1 × 104 cells/well) according to the manufacturer’s instructions and protocols. The calibration plate and probe plate were then carefully inserted into an Agilent Seahorse Bioscience XF96 extracellular flux analyzer, where the OCR was precisely calculated using the Wave software provided by Agilent.
Biochemical analyses
The blood samples were centrifuged (5000 rpm) for 15 min at 4 °C to collect the serum for further biochemical measurements. According to the corresponding manufacturer’s instructions, the concentration levels of cTnI at 0.5, 3, and 6 days after surgery were consecutively detected using a standard enzyme-linked immunosorbent assay kit (#E-EL-M1203, Elabscience, China), as well as the LDH levels at 6 days after surgery (LDH Assay Kit, # MAK066, Beckman Coulter, Brea, CA, USA). All measurements were performed in duplicate.
Magnetic resonance imaging protocol
For further assessing the alterations of cardiac structures and functions after I/R injury, cardiac magnetic resonance imaging (cMRI) was performed at 3 days after surgery. A dedicated small-animal MRI system (BioSpec70/20, Bruker, Germany) equipped with AVANCE III electronics and Paravision 6.0 software was used as previously described [25]. All mice were anesthetized via inhaling 3% isoflurane initially and maintained with 1.5% isoflurane during examinations. After standard cardiac localization, cine images were acquired and reconstructed using electrocardiogram-triggered fast low-angle shot sequences in four or five short-axis planes encompassing the entire left ventricle. All images were analyzed using dedicated software (QMass MR v.7.6, Medis, Leiden, The Netherlands, and ImageJ software). Edema size was quantified from the extent of hyperintense areas on T2-weighted images and calculated as a proportion of the total left ventricular area.
Real-time fluorescence quantitative polymerase chain reaction (RT-qPCR) analysis
To measure the expression levels of the associated genes, heart samples were analyzed via RT-qPCR. Total RNA was extracted using TRIzol reagent (R411-01, Vazyme, China), then transcribed into complementary DNA (cDNA) using the HiScript II Q Select SuperMix (R233-01, Vazyme, China). The amplification of cDNA was subsequently performed following the protocol (95 °C for 30 s, followed by 40 cycles at 95 °C for 5 s, 60 °C for 20 s) of the SYBR qPCR Master Mix (Q711-02, Vazyme, China) using the iCycler iQ system (Bio-Rad). The expressions of purine nucleoside phosphorylase (PNP) and cytosolic purine 5′-nucleotidase (NT5C2) were measured, and 18S was chosen as an internal control. The primer sequences of relative genes were as follows: 18S: 5′-GGAGTTCCTTGCGATGGTGA-3′, 3′-GGCCAGAAGCTCTGCTACTC-5′; PNP: 5′-GTGTGTGCGACACCGTACT-3′, 3′-CTCGGCTAGGTAGCGGTAG-5′; NT5C2: 5′-TCAAAGAAACGGCAAGGGTGGAG-3′, 3′-GCTGTCCAGGTGCTTGTAGAGTTC-5′. All samples were analyzed in duplicate, and the comparative Cq method (2-ΔΔCq) was used for quantification.
Western blot analysis
Protein homogenates were extracted from heart tissues and NRCMs using radioimmunoprecipitation assay (RIPA) lysis buffer (C1053, Applygen, China). A bicinchoninic acid (BCA) protein assay kit (BL521A, Biosharp, China) was used to detect their concentrations. Thereafter, protein samples (20–40 μg) were separated on 8–12% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) as appropriate and transferred to a polyvinylidene fluoride (PVDF) membrane. After blocking in 5% skim milk for 1–2 h at 37 °C, these membranes were incubated with the specific primary antibodies. Detection was performed using enhanced chemiluminescence system (BL521A, Biosharp, China), and the intensity of the protein bands was analyzed using ImageJ software. The primary antibodies in this study were as followed: GPX4 (#ab125066, Abcam), ACSL4 (#ab155282, Abcam), NT5C2 (#15223-1-AP, Proteintech), PNP(#A19885, Abclonal), BAX (#50599-2-Ig, Proteintech), PHGDH (#YP-mAb-12503, UpingBio), GAA (#YP-mAb-12503, UpingBio), XPP1(#YP-mAb-06825, UpingBio), ALDH1A(#YP-mAb-11363, UpingBio), EPHB3 (#YP-mAb-12915, UpingBio), and UMPS (#14830-1-AP, Proteintech). The expression of GAPDH (#60,004–1-Ig, Proteintech) or HSP90 (#13171-1-AP, Proteintech) was chosen as an internal control as appropriate.
Evaluation of mitochondrial morphology
The electron microscopy was used for assessing the mitochondrial morphology in these NRCMs. After fixing with 2.5% glutaraldehyde at room temperature for 5 min, these cells were collected (a cell scraper was used to scrap these cells at a single direction to avoid repeated damage) and then centrifuged at 1000 rpm for 2 min. We gently resuspended the cells in an adequate volume of electron microscope fixative, then sectioned the fixed cells and examined them under the electron microscope. Mitochondrial damage was assessed utilizing the Flameng scoring system.
Measurement of intracellular Fe2+
Intracellular Fe2⁺ levels were detected using the FerroOrange fluorescent probe (F374, Dojindo, Kumamoto, Japan) according to the manufacturer’s instructions. Briefly, the NRCMs were cultured in conditioned medium in confocal dishes. Next, the cells were rinsed and incubated with 250 µM FerroOrange for 30 min. Afterwards, the cells were observed under a confocal microscope (FV3000, Olympus, Japan) and analyzed using FV3000 and ImageJ software.
Lipid peroxide measurement
Lipid peroxides were measured using the Liperfluo fluorescent probe (L248, Dojindo, Kumamoto, Japan) according to the manufacturer’s instructions. An inverted fluorescence microscope was used for further observation, and the acquired images were analyzed using ImageJ software.
Statistical analysis
All results are presented as mean ± standard deviation (SD), and GraphPad Prism software (version 8.0, GraphPad Software, Inc., CA, USA) was applied for all analyses. Comparisons between multiple groups were conducted using one-way or two-way analysis of variance (ANOVA). A two-tailed P-value <0.05 or Q-value <0.05 (false discovery rate (FDR)-adjusted) was considered statistically significant.


Comments (0)