Human tissue harvest and preparation
All donors were patients in the School of Stomatology, The Fourth Military Medical University, and have signed informed consent to this study. Experimental procedures of human samples were approved by the Ethics Committee of The Fourth Military Medical University with the approval number IRB-REV-2022187. Dental follicles were harvested based on inclusion criteria as follows: patients were aged 20 ± 2 years old, had no history of pain or infection, and had unerupted third molars with healthy surrounding tissues. Dental papilla tissues were acquired from the third molars that had not completely developed roots with open apical foramen. All the patients were examined by oral panorama before surgery, and any of the third molars with an abnormal low-density shadow around were excluded. After local anesthesia, incising and flap elevation were performed to expose the bone covering the teeth. Fissure burs assembled in a high-speed drill were used to remove a portion of bone and provide space for elevator usage. After tooth extraction, the wound was closed carefully, and patients were given instructions for post-operative care. Dental tissues attached to the teeth were immersed in 10% alpha-minimum essential medium (α-MEM; 12571-048, Invitrogen, USA) and immediately delivered to the laboratory in an ice box. Dental follicles were obtained by cutting off soft tissues around teeth, and dental papilla were isolated by amputation of soft tissues out of the root apex. The tissues were sectioned into 2 mm³ pieces and rinsed twice with phosphate-buffered saline (PBS; P5493, Sigma-Aldrich, USA) before undergoing the following experiments.
ScRNA-seq analysis
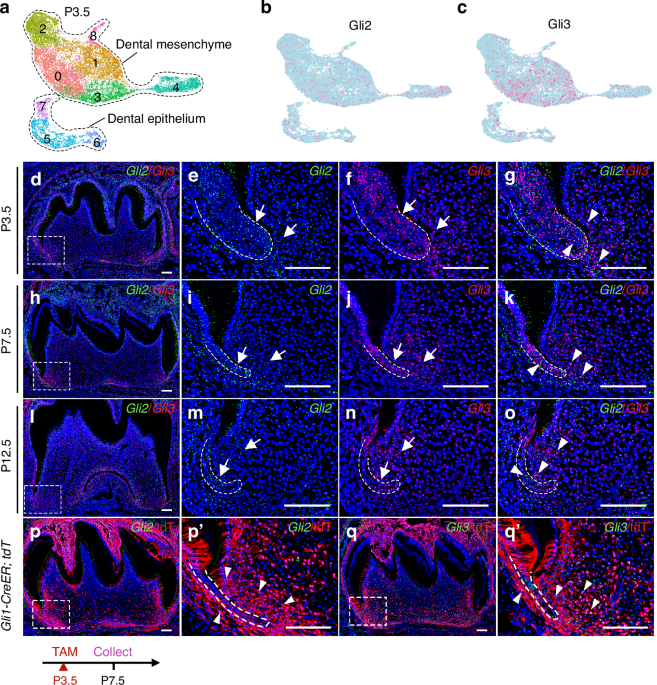
Dental tissues were initially dissociated into single cells using 0.02% Type I collagenase (17018029, Gibco, USA). Cells were barcoded with 10× gel beads and encapsulated in oil to form single-cell gel beads-in-emulsion (GEMs). Reverse transcription reactions were engaged in barcoded full-length cDNA, followed by the disruption of emulsions using the recovery agent. Single-cell 3′ Reagent v3 Kits (1000268, 10× Genomics, USA) were used for scRNA-seq library construction. The sequencing was performed on the Illumina Nova 6000 PE150 platform (Illumina, USA). Cell Ranger (version 7.0.1) software (10× Genomics, USA) was employed for quality control and through comparisons between reads to the genome by the spliced transcript alignment to a reference aligner. The cells that met the criteria were retained: (1) gene numbers > 200, unique multiplex index (UMI) > 1 000, and log10GenesPerUMI > 0.7; (2) the UMI of mitochondrial genes < 15% and hemoglobin genes < 5%. To mitigate batch effects, mutual nearest neighbor (MNN) analysis was conducted. Cells were then clustered using the MNN clustering algorithm and visualized using the two-dimensional uniform manifold approximation and projection (UMAP) algorithm. For a detailed identification of cell types, homotypic clusters were selected for re-analysis, graph-based clustering, and marker analysis. The top 100 genes of DFSCs and SCAP, ranked according to gene differences, were analyzed and displayed as a Venn plot performed on the OECloud tools at https://cloud.oebiotech.com. The dot plots, feature plots, and violin plots of genes were also performed on the OECloud tools at https://cloud.oebiotech.com. The violin plots of gene expression level were drawn using ggplot2 in the R package (4.0.3). GO and KEGG enrichment analysis was performed using ClusterProfiler of the R package, and an adjusted P value < 0.05 was set as a limitation (ListHitså 2). RNA velocity analysis was performed using the Python script velocyto.py on the Cell Ranger output folder to elucidate the dynamics of cell subpopulations. The results were projected to UMAP and visualized using Seurat software. PDGFRA+ DFSCs were counted by threshold > 0. Cell-cell communication was conducted using CellChat (version 1.1.3) to examine interactions involving ligands, receptors, and their cofactors. After standardizing of expression matrix and creating cellchat objects, preprocessing was performed using default parameters. Potential ligand–receptor interactions were calculated, and the aggregateNet function was utilized to achieve the intercellular communication networks aggregated.
Cell cultures
Dental tissues were obtained as mentioned above, cut into pieces, and digested with 0.02% Type I Collagenase for 1 h (17018029, Gibco, USA). Primary DFSCs and SCAP were cultivated in α-MEM supplemented with 10% fetal bovine serum (FBS; Gibco, USA), 2 mmol/L L-glutamine (35050061, Invitrogen, USA), 100 μg/mL penicillin, and 100 IU/mL streptomycin (15070063, both from Invitrogen, USA).
HUVECs were sourced from iCell Bioscience Inc. (Shanghai, China) and maintained in an EC medium (1001, ScienCell, USA). Cells were passaged using a 0.25% trypsin solution (15050057, Gibco, USA) as they reached 80%–90% confluence. Culture conditions were maintained at 37 °C in a 5% CO2 atmosphere, with medium changes every 3 days.
Flow cytometric analysis
Cultured DFSCs and SCAP were harvested and stained with an anti-human CD90-PE antibody (5 µL per test, 12-0909-41, eBioscience, USA), CD90-FITC antibody (5 µL per test, 11-0909-42, eBioscience, USA), and an anti-human CD45-PE antibody (5 µL per test, 304058, Biolegend, USA), respectively. Samples were incubated at 4 °C in the dark for 1 h, filtered, and prepared for analysis using a flow cytometer (CytoFLEX, Beckman, USA). To validate the proportion of PDGFRA+ cells in dental follicle tissue cells and cultured DFSCs, cells were stained with an anti-human PDGFRA-PE antibody (5 µL per test, sc-398206 PE, Santa Cruz Biotechnology, USA) and were analyzed using a flow cytometer (CytoFLEX, Beckman, USA) after 1-h incubation at 4 °C in the dark.
CFU assay
DFSCs and SCAP were plated and cultured in 6-well dishes (Corning, USA) at a density of 1000 cells per well. After 14 days, cells were fixed with 4% paraformaldehyde (PFA; 1004965000, Sigma-Aldrich, USA) and stained with 1% crystal violet (C8470, Solarbio, China) for 5 min. The experiment was performed thrice to ensure the reliability and consistency of results.
EdU assay
DFSCs and SCAP were seeded in 24-well plates (Corning, USA) at 1 × 104 cells per well and incubated for 24 h. Cells were then treated with EdU at a working concentration of 10 μmol/L in 200 μL of culture medium for 48 h. Afterward, the cells were detected using a VF 488 Click-iT EdU universal cell proliferation detection kit (HY-K1087, MCE, China) according to the manufacturer’s instructions.
Osteogenic and adipogenic differentiation
DFSCs and SCAP were seeded in 6-well plates at a density of 3 × 105 each per well. The culture medium was replaced with the osteogenic medium or the adipogenic medium until full confluence was achieved. The osteogenic medium was prepared, consisting of α-MEM supplemented with 10% FBS, 5 mmol/L β-glycerophosphate (G5422, Sigma-Aldrich, USA), 10 nmol/L dexamethasone (D4902, Sigma-Aldrich, USA), and 50 μg/mL ascorbic acid (02100769-CF, MP Biomedicals, USA). The adipogenic medium was prepared consisting of α-MEM supplemented with 10% FBS, 200 μmol/L indomethacin (I8280, Sigma-Aldrich, USA), 2 mmol/L insulin (I3536, Sigma-Aldrich, USA), 0.5 mmol/L isobutylmethylxanthine (IBMX; I5879, Sigma-Aldrich, USA), and 10 nmol/L dexamethasone (D4902, Sigma-Aldrich, USA). The medium was refreshed every 3 days. After induction for 14 days, cells were fixed with 4% PFA for 30 min. Mineralized nodules were stained with a 1% alizarin red S solution (60504ES25, Yeasen, China), and lipid droplets were stained with a 0.3% oil red O solution (O104972, Aladdin, China) at room temperature for 20 min. After rinsing with PBS twice, photographs were captured under a bright field of fluorescence microscope (Olympus, Japan) or using an inverted microscope (Leica, Germany). For the detection of ALP activity, supernatants from cell cultures of each group were collected, and ALP activity was detected using an ALP activity assay kit (A059-2, Nanjingjiancheng, China) according to the manufacturer’s instructions. The absorbance of each well was determined by measurements at 520 nm.
Magnetic cell sorting
DFSCs of the first passage were used for sorting. Single-cell suspension was prepared and incubated with 2 μL of a biotinylated CD140a (i.e., PDGFRA) monoclonal antibody (13-1401-80, Themofisher, USA) at room temperature for 10 min. Cells were then conjugated with MagniSort™ Streptavidin Positive Selection Beads (MSPB-6003-74, Themofisher, USA) according to the manufacturer’s instructions. After being washed with the separation buffer, cells were subjected to a magnetic field to separate positive and negative populations.
ELISA
The conditioned medium was collected and centrifuged at 4 °C for 20 min at a speed of 3 000 r/min. The VEGFA concentrations were measured using a commercial ELISA kit (F111309-A, FANKEW, China) according to the manufacturer’s instructions.
qRT-PCR
Total RNA was extracted using the MIsZOL Reagent (MI00617, Mishushengwu, China). 5× PrimeScript RT Master Mix (RR036A-1, Takara, Japan) was used for cDNA synthesis, and reverse transcription proceeded on the PCR Amplifier (EasyCycler, Germany). qPCR was performed using TB Green® Premix Ex Taq (RR420A, Takara, Japan), and gene expression was detected using a Real-Time System (Bio-Rad, USA). The CFX Manager software (Bio-Rad, USA) was utilized for result analysis. The primer sequences (5′–3′) were listed as: h-PDGFRA forward: AAGAGATCATTGGAGGCCGTG; reverse: AGGATTAGGCTCAGCCCTGT; h-CCND2 forward: GGTCATCCTTGGTCTATGTGCTCTG; reverse: GGGTTGTCTTCTCCTCTGGCTTTG; h-CCND3 forward: ACGAGGAGGTATGTGAGGAGCAG; reverse: AGACAGGTAGCGATCCAGGTAGTTC; h-CDK2 forward: AGGATGTGACCAAGCCAGTACCC; reverse: CCACCTGAGTCCAAATAGCCCAAG; h-CDK3 forward: AGGAGGGAGTGAGGGAGAGGAG; reverse: AGCACATCTCAGGTGAAGGAACAAC; h-ALP forward: TAAGGACATCGCCTACCAGCTC; reverse: TCTTCCAGGTGTCAACGAGGT; h-RUNX2 forward: CTTTACTTACACCCCGCCAGTC; reverse: AGAGATATGGAGTGCTGGTC; h-OCT4 forward: GCCGTATGAGTTCTGTGGGG; reverse: CTCCTTCTCCAGCTTCACGG; h-SOX2 forward: ACACCAATCCCATCCACACT; reverse: GCAAACTTCCTGCAAAGCTC; h-NANOG forward: TGAACCTCAGCTACAAACAG; reverse: CTGGATGTTCTGGGTCTGGT; h-GAPDH forward: CTTTGGTATCGTGGAAGGACTC; reverse: GTAGAGGCAGGGATGATGTTCT.
Cell migration assay
HUVECs were seeded into 6-well plates at a density of 5 × 105 cells per well. At 90% confluence, scratches were created using sterile pipette tips. Conditioned medium from DFSCs was collected, filtered through 0.22 µm filters, and mixed with fresh and serum-free EC medium at a 1:1 ratio before addition. Scratch areas were photographed at 0, 12, and 24 h using an inverted microscope (Leica, Germany) and quantified using the ImageJ software (NIH, USA).
Tube formation assay
Capillary-like network formation was assessed through an in vitro tube formation assay on μ-Slides 15 Well 3D (81506, ibidi, Germany), precoated with Matrigel (082703, Shanghai Nova Medical Technology, China). HUVECs were seeded at 1 × 104 cells per well. Conditioned medium from DFSCs was collected, filtered through 0.22 µm filters, and mixed with fresh and serum-free EC medium at a 1:1 ratio before addition. Tube formation was observed and photographed after 5 h using an inverted microscope (Leica, Germany), and the number of network structures was quantified using the ImageJ software (NIH, USA).
PDGFBB treatment
All experiments involving DFSCs treated with PDGFBB were conducted by supplementing the culture medium with PDGFBB (HY-P7055, MCE, China) at a concentration of 100 ng/mL when the cell confluence reached 40%-50%. After 48 h, conditioned medium was collected for indirect coculture with ECs and deposited at −80 °C. Additionally, the DFSCs were applied for qRT-PCR and western blot analysis. As for osteogenic differentiation assays, the medium was refreshed along with PDGFBB at the same concentration of 100 ng/mL.
Aggregate culture
For the induction of cell aggregates, DFSCs were seeded into 24-well plates at a density of 1 × 104 cells per well. Once cells reached 80% confluence, the medium was replaced with α-MEM supplemented with 50 μg/mL ascorbic acid (02100769-CF, MP Biomedicals, USA), 2 mmol/L L-glutamine (35050061, Invitrogen, USA), 100 μg/mL penicillin, and 100 IU/mL streptomycin (15070063, both from Invitrogen, USA). The medium was refreshed every 2 days. After ~7 days, cell aggregates with white membrane-like structures were formed and were acquired from the culture plates using a cell scraper.
Live/Dead cell staining
Aggregates induced in the 6-well plate were washed with PBS twice. According to the kit instructions (C2015M, Beyotime, China), the staining working solution was added and incubated at 37 °C in the dark for 30 min. Subsequently, counterstain the nuclei with DAPI. After washing with PBS, observe and capture images under a fluorescence microscope.
Rats
Fifty male Sprague-Dawley rats, aged 8 weeks with weighing 200–220 g, were provided by the Laboratory Animal Centers of The Fourth Military Medical University. The animals were housed in a well-ventilated environment with a standard 12-h light-dark cycle and free access to water and food. All animal experiments were performed in compliance with relevant laws and ethical regulations, following ARRIVE guidelines, and approved by the Ethics Committee of The Fourth Military Medical University with the approval number IRB-REV-2022187.
Rats were anesthetized using 3% isoflurane via an anesthesia machine (RWD, China), and the right mandibles were exposed. A box-type periodontal window defect (3 × 2 × 1 mm3 in width, height, and depth, respectively) at the buccal side of the first molar was surgically created using a 0.8 mm diameter drill. Following the establishment of defects, DFSC aggregates were implanted, and defects without any implantation served as blank controls. At 6 weeks post-surgery, the animals were euthanized using CO2, and their mandibles were harvested for further analysis.
Micro-CT analysis
For quantitative assessment of periodontal bone regeneration, the right mandibles were meticulously excised and fixed in 4% PFA after the removal of soft tissues. The specimens were then prepared for micro-CT scanning using a PerkinElmerTM Quantum GX2 micro-CT device (PerkinElmer, USA) at the Animal Center of The Fourth Military Medical University. Scanning parameters included an energy setting of 90 kV and a current of 80 μA, with a resolution of 18 μm. The acquired data were processed for 3D reconstruction using the VG Studio MAX software (version 2023.2.1) (Volume Graphics, Germany). Bone volume over tissue volume (BV/TV) was calculated to provide a quantitative analysis of the regenerated bone.
Histological analysis
Dental follicles, dental papilla, and cell aggregates were fixed in 4% PFA for 12 h. Rat mandibles were harvested and fixed in a 4% PFA for 24 h, followed by decalcification in 17% ethylenediaminetetraacetic acid (EDTA) for 4 weeks. The dental follicle, dental papilla, cell aggregates, and decalcified samples underwent a series of ethanol and xylene for dehydration before being embedded in paraffin. Sections of 5 μm thickness were meticulously prepared using a microtome (Leica, USA). H&E staining and Masson’s staining were performed according to established protocols.64,72 Images were taken by the SLIDEVIEW VS200 (Olympus, Japan).
IF staining
For cultured DFSC and SCAP, cells were washed twice with PBS and fixed with 4% PFA at room temperature for 30 min. Fixed cells were permeabilized with 0.5% Triton X-100 (X100PC, Sigma-Aldrich, USA) for 15 min. Cells were then blocked in 5% bovine serum albumin (BSA; 0881066-CF, MP Biomedicals, USA) in PBS for 30 min at room temperature. Cells were stained with primary antibodies for Nestin (sc-33677, Santa Cruz Biotechnology, USA; diluted 1:100), CK14 (ab7800, Abcam, UK; diluted (1:100), and PDGFRA (sc-398206 PE, Santa Cruz Biotechnology, USA; diluted 1:100), overnight at 4 °C, and then washed three times with PBS. Secondary antibodies were stained for 1 h at room temperature and then washed three times with PBS before DAPI staining (ab104139, Abcam, UK) for 5 min. Cells were examined under a fluorescence microscope (Olympus, Japan).
For HUVECs, after tube formation on the µ-Slides, capillary-like structures were fixed with 4% PFA for 30 min, followed by rinsing with PBS and blocking in 5% BSA for 30 min at room temperature. Then, HUVECs were incubated with primary antibodies for CD31 (FAB3628G, R&D Systems, USA; diluted 1:100) and EMCN (DF13357, Affinity, China; diluted 1:100) at 4 °C overnight. Fluorescence-conjugated secondary antibodies were then stained at 37 °C for 1 h, followed by DAPI staining for 10 min. The fluorescent images were captured with a confocal laser scanning microscope (CLSM) (Nikon, Japan). The percentage of CD31+EMCN+ vessels was analyzed by the ImageJ software (NIH, USA).
For PFA-fixed human dental follicles, dental papilla, and decalcified mouse mandibles, tissues were dehydrated in 30% sucrose solution (Sigma-Aldrich, USA) and embedded in the O.C.T compound (4583, Sakura Finetek, USA) to obtain 10 μm frozen sections in a freezing microtome (Leica, Germany). After permeabilization with 0.3% Triton X-100 (X100PC, Sigma-Aldrich, USA) for 15 min and blocking with the goat serum (AR0009, BOSTER, China) for 30 min at room temperature, sections were incubated with primary antibodies for PDGFRA (sc-398206 PE, Santa Cruz Biotechnology, USA; diluted 1:100), MSX1 (bs-8512R, Bioss, China; diluted 1:100), PAX9 (A19741, ABclonal, China; diluted 1:100), RUNX2 (sc-390715, Santa Cruz biotechnology, USA; diluted 1:100), CD31 (FAB3628G, R&D Systems, USA; diluted 1:100), EMCN (DF13357, Affinity, China; diluted 1:100), rat PDGFBB (ab21234, Abcam, UK; 1:100), and human VEGFA (ab9570, Abcam, UK; 1:100) at 4 °C overnight. Fluorescence-conjugated secondary antibodies were then stained at 37 °C for 1 h, followed by DAPI staining for 10 min. The fluorescent images were captured with CLSM (Nikon, Japan).
Western blot analysis
Proteins were extracted on ice using a RIPA buffer (P0013C, Beyotime, China) supplemented with protease inhibitors for 10 min. After protein quantification using a BCA assay (PA115-02, TIANGEN, China), 20 μg proteins were loaded onto sodium dodecyl sulfate-polyacrylamide gels and underwent electrophoresis, followed by transferred onto polyvinylidene fluoride membranes (GVWP02500, Roche, Switzerland) and blocked in 5% BSA (0881066-CF, MP Biomedicals, USA) for 1.5 h at room temperature. The membranes were incubated overnight at 4 °C with primary antibodies diluted in PBS containing 0.1% Tween 20 (PBST). Then membranes were washed 3 times with PBST and incubated at room temperature with secondary antibodies for 1.5 h. After further washing with PBST, the protein bands were visualized with an enhanced chemiluminescence kit (4AW011-100, 4A Biotech, China) and evaluated with a gel imaging system (4600, Tanon, China). The primary antibodies used were: anti-COLI antibody (ab260043, Abcam, UK; diluted (1:1 000), anti-OSX antibody (ab209484, Abcam, UK; diluted 1:1 000), anti-OPN antibody (22952-1-AP, Proteintech, China; diluted 1:1 000), anti-RUNX2 antibody (12556, Cell Signaling Technology, USA; diluted 1:1 000), anti-PDGFBB antibody (ab16829, Abcam, UK; diluted 1:1 000), anti-p-PI3K antibody (AF3242, Affinity, China; diluted 1:1 000), anti-PI3K antibody (AF5112, Affinity, China; diluted 1:1 000), anti-p-AKT antibody (AF0016, Affinity, China; diluted 1:1 000), anti-AKT antibody (ab179463, Abcam, UK; diluted 1:1 000), anti-p-mTOR antibody (5536T, Cell Signaling Technology, USA; diluted 1:1 000), anti-mTOR antibody (2983T, Cell Signaling Technology, USA; diluted 1:1 000), anti-NICD antibody (3608S, Cell Signaling Technology, USA; diluted 1:1 000), and anti-GAPDH antibody (30201ES20, Yeasen, China; diluted 1:2 000). The secondary antibodies included Peroxidase-AffiniPure Goat Anti-Mouse IgG (H+L) (115-035-003, Jackson ImmunoResearch, USA) and Peroxidase-AffiniPure Goat Anti-Rabbit IgG (H+L) (111-035-003, Jackson ImmunoResearch, USA), and the secondary antibody dilute ratio was 1:10 000.
Statistical analysis
All data were presented as mean ± standard deviation (SD). The normality and variances of all data were evaluated before comparisons, and all data matched the normal distribution. As for two samples, comparisons were made using unpaired two-tailed Student’s t-tests for data with equal variances, and Welch’s correction was employed when equal variances were not assumed. Multiple samples were compared using ordinary one-way analysis of variance (ANOVA) with Tukey’s post-hoc tests when equal variances were assumed. Brown-Forsythe and Welch ANOVA tests with Games-Howell’s multiple comparisons tests were employed when equal variances were not assumed. For all experiments, P < 0.05 was considered to be significant. The GraphPad Prism software (Version 8.0.1) was used for all statistical analysis.



Comments (0)