
Remember me


Briefly, hiPSCs (WTC-11 human induced pluripotent stem cells) (Coriell, #GM25256) were seeded on 12-well plates coated with growth factor-reduced Matrigel (Corning, #356231) and cultured in mTeSR medium (StemCell Technologies, #85850) until cells reach confluency with medium changes daily. On the first day of differentiation (Day 0), stem cell media is replaced with base iAM media consisting of EpiCult-C media (StemCell Technologies, #05630) supplemented with 1× EpiCult-C Proliferation Supplement (STEMCELL Technologies), 0.1 μmol/L β-mercaptoethanol (BME) (Sigma, #M7522), 1× Hydrocortisone Stock Solution (96 μg/mL), 0.05X GlutaMAX (Gibco), 0.4× NEAA (Gibco) and 1× Penicillin-Streptomycin (Gibco) and 400 nmol/L smoothened agonist (SAG) (Selleckchem, # S7779). On day 3 of differentiation 150 pmol/L of monomeric bone morphogenic protein-4 (BMP4) (rndsystems, #314-BP-010) is added daily until day 7. At day 8, the base media is supplemented with 1 μmol/L of BMP-I inhibitor (LDN-193189) (Tocris, #6053), 5 μmol/L of GSK3-Inhibitor (CHIR99021) (Selleckchem, #4423), 500 pmol/L epidermal growth factor (EGF) (rndsystems, #236-EG) and 70 pmol/L of Neurotrophin-4 (NT4) (rndsystems, #268-N4). No media refresh is added on day 9. On day 10, repeat with the same small molecules used on day 8. From day 12 onwards the cultures were then extended until day 16 by adding 1× N2 Supplement (Gibco #17502-048), 400 nmol/L SAG, 300 pmol/L BMP4, 5 μmol/L of GSK3-Inhibitor (CHIR99021), 500 pmol/L of EGF 2 nmol/L transforming growth factor beta 1(TGFβ1) (rndsystems, #7754-BH) 70 ppmol/L of Neurotrophin-4 (NT4) (rndsystems, #268-N4) and 300 pmol/L of Activin (Peprotech, #120-14 P) and for the early ameloblast stage at day 16. The media containing the small molecules were changed every other day from day 12 until day 16.
Production and characterization of the C3-DLL4 notch activatorThe C3-DLL4 Notch activator was produced using a previously established computationally designed scaffold with defined valency and geometry.23 The C3 scaffold, a homotrimeric helical bundle, was conjugated to DLL4 using SpyLigation, which forms a stable isopeptide bond between SpyCatcher (SC) and SpyTag (ST). The C3-SpyCatcher fusion was expressed in Escherichia coli, while DLL4-SpyTag was produced in mammalian Expi293F cells, ensuring proper folding and post-translational modifications. Both components were purified using affinity chromatography, and successful conjugation was confirmed by SDS-PAGE (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) and Coomassie staining, showing a higher molecular weight band corresponding to the assembled C3-DLL4 complex. For cell treatment, a 10 µM stock solution of C3-DLL4 was prepared by conjugating DLL4-ST with C3-SpyCatcher, followed by serial dilutions to achieve precise ligand presentation and receptor engagement.
Notch activation assay in reporter cell lineThe Notch activation assay was performed as described before23 using U2OS-N1-Gal4/UAS-H2B-mCitrine cells49,50 to evaluate the effect of C3-DLL4 on Notch signaling. Cells were seeded in a 96-well tissue culture-treated plate at a density of 15 000 cells per well in DMEM supplemented with 10% FBS, 1× Penicillin-Streptomycin, 0.5X NEAA (Gibco), 1× GlutaMax, and 1× Sodium Pyruvate. Doxycycline (Dox, 1 µg/mL) was added at seeding to induce Notch1-Gal4 (N1-Gal4) expression, which is essential for the assay. On Day 1, cells were treated with C3-DLL4. Through serial dilutions of C3-DLL4 the final concentrations of 200, 100, 50, 10 and 1 nmol/L (DLL4 molar equivalent) were added to respective wells in replicates. Untreated wells served as negative controls. After treatment, the cells were gently mixed and incubated under standard culture conditions at 37 °C with 5% CO₂. On Day 2, the media was replaced with fresh Dox-containing media to maintain Notch induction. On Day 4, the Notch activation was assessed by measuring H2B-mCitrine fluorescence. Fluorescence intensity was quantified to determine the dose-dependent activation of Notch signaling. Control wells without C3-DLL4 were analyzed to establish baseline fluorescence, ensuring accurate interpretation of Notch activation levels.
iPSC derived odontoblast differentiationWe modified the odontoblast differentiation protocol previously described by our lab8,26 to avoid serum usage and developed serum-free protocol for the OB differentiation. This differentiation protocol begins with hiPSCs, treated with dual SMAD inhibitors, SB431542 and LDN-193189 (Tocris, #6053), to inhibit TGF-β and BMP signaling pathways, respectively. This promotes ectodermal lineage commitment, guiding the hiPSCs toward an odontogenic trajectory. From Day 0 to Day 11, the WNT signaling pathway is activated using CHIR99021, a GSK-3β inhibitor, to direct mesenchymal lineage specification and to facilitate the generation of neural crest cells (iNCs), a precursor population essential for odontoblast development. On Day 12, BMP4 is introduced to drive the differentiation of iNCs into odontoblast precursors in serum-free conditions.
Specifically, p75 + iNC cells were cultured in Serum-free (SFM) Odontogenic Medium, consisting of DMEM + Glutamax (Gibco 10566016), 100 nmol/L dexamethasone (Sigma-Aldrich D4902), 15% KnockOut Serum Replacement (KOSR), 0.5X NEAA (Gibco), 0.005 mM ITS-A (Gibco 51300-044), 5 mmol/L β-glycerophosphate (Sigma-Aldrich G9422), and 50 μg/mL L-ascorbic acid (Sigma-Aldrich #A4544) for 14 days (OB). Odontogenic Medium was supplemented with 50 ng/mL BMP4 (Stemcell Technologies #78211) for Day 11 to Day 18, followed by 25 ng/mL BMP4 (Stemcell Technologies #78211) and 400 nmol/L SAG (Stemcell Technologies #73412) from Day 18 until Day 25 supplemented with 100 ng/mL C626,29 for 14 days (iOB C6); followed by treatment with or without Notch activator (50 nmol/L C3-DLL4)26 (iOB C6 N) on Day 18 for 24 h followed by change of media on Day 19 followed until Day 25. All cultures were performed on Matrigel-coated plates at a 1:30 dilution and incubated at 37 °C with 5% CO2. Each differentiation was performed in triplicate, with undifferentiated hiPSC as the negative control.
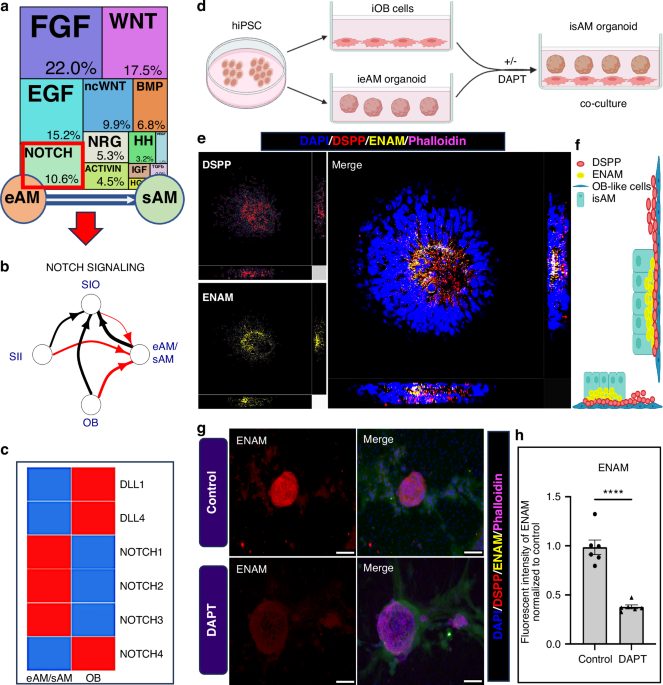
Establishing a functional co-culture model for ameloblast and odontoblasthiPSCs were differentiated into ameloblast and odontoblast lineages to establish a functional co-culture model for ameloblasts and odontoblast lineages under defined serum-free conditions. For ameloblast differentiation, hiPSCs were cultured in a 2D monolayer to generate early ameloblasts (ieAM) from Day 0 to Day 16. On Day 16, differentiated iAM cells were trypsinized using TrypLE Express (Thermo Scientific or Gibco, #12604013). The cells were gently resuspended and then transferred into 24-well ultra-low attachment plates for three-dimensional (3D) suspension culture in Epicult Plus medium (Stemcell Technologies #06070), allowing them to self-organize into ieAM organoids. The ieAM organoid cultures were incubated at 37 °C with 5% CO2, and the media were changed every other day. These organoids were cultured until the next 7 days, stabilizing their maturation. Concurrently, odontoblast cells were made similarly in a separate plate by culturing iOBs differentiated from iNCs (neural crest) that were differentiated from hiPSCs in a serum-free odontogenic differentiation medium. The differentiated iOB cells were plated as monolayer mixed in 25% (v/v) of Matrigel (Corning, #356231) diluted in odontogenic media in a glass-bottomed 24-well plate (Corning, #3603). The next day, ieAM organoids suspended in the ameloblast base medium and 10 μM ROCKi (Y-27632, Selleckchem, #S1049) were added on top of the iOB monolayer and then incubated for 24 h at 37 °C in 5% CO2. The co-culture was supplemented with fresh media (1:1 mixture of ameloblast and odontogenic media) every three consecutive days. The co-culture was collected on Day 37 for further analysis.
DAPT inhibition assayTo assess the effect of Notch inhibition on ameloblast-odontoblast interactions, a DAPT inhibition assay was performed after 4 days of the co-culture (Day 29). DAPT (10 μmol/L) (SantaCruz Biotech #sc-201315) was added to the co-culture medium to inhibit Notch signaling, and the treated co-culture was incubated for 24 h at 37 °C with 5% CO₂. After 24 h, the media was replaced with a fresh co-culture medium to continue differentiation. The co-culture was maintained under standard conditions and collected on Day 37 for further analysis of ameloblast maturation and enamelin secretion.
Notch activation in the co-culture assay using C3-DLL4Notch activation was induced in the co-culture system by treating ieAM organoids with C3-DLL4 (50 nmol/L) after 4 days of co-culture on Day 29. The treatment was maintained for 24 h, after which the media was replaced on Day 30. Co-cultures were maintained with media changes every 3 days until Day 37, when they were collected for further analysis.
Pathway analysis (TopPath)To reanalyze the pathway involved in ameloblast maturation, we used TopPath pipeline, as described previously with minor changes. In brief, the talklr R package27 was used to pinpoint ligand-receptor interactions specific to each cell type during the transition stage of ameloblast maturation from early ameloblast to secretory ameloblast. The DEsingle51 and scMLnet52 tools were applied to analyze downstream signaling by creating multilayer networks that connect ligands to receptors and transcription factors to their corresponding differentially expressed target genes. Pathway activity scores were then calculated, reflecting the percentage (0–100%) of total activity across all pathways evaluated in the analysis. The pathways considered in the analysis were: TGFβ, BMP, GDF, GDNF, NODAL, ACTIVIN, WNT, ncWNT, EGF, NRG, FGF, PDGF, VEGF, IGF, INSULIN, HH, EDA, NGF, NT, FLT3, HGF, NRXN, OCLN, NOTCH.
Generation of DLX3 KO iPSCOne million WTC11 iPSC were electroporated with Cas9 (0.3 μmol/L, Sigma) and gRNA targeting DLX3 (1.5 μmol/L, Synthego) as RNP complex using Amaxa nucleofector (Human Stem Cell kit 2) in the presence of ROCK inhibitor. Individual colonies were hand-picked and plated into 96-well plates. DNA was extracted using Quick Extract DNA extraction solution (Epicentre #QE09050), and nested polymerase chain reaction (PCR) was performed using Phusion Flash polymerase (ThermoFisher, #F631S). The PCR product was purified using ExoSap-IT (Thermofisher) and sent for Sanger sequencing analysis (Genewiz, Azenta Life Sciences) to identify potential KO clones. gRNA sequence: TAGCTGGAGTAGATCGTACG. PCR primer sequences: F: GAAGGCGTCGTGAGCGAAG, R: TAGCCTGGAGGGAAAACACG. KO was verified at the protein level after 14 days of odontoblast differentiation and 16 days of ameloblast differentiation.
Confirming knockout at the protein levelCells were lysed directly on the plate using a lysis buffer containing 20 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 15% glycerol, 1% Triton X-100, 1 mol/L β-glycerophosphate, 0.5 mol/L NaF, 0.1 mol/L sodium pyrophosphate, orthovanadate, PMSF, and 2% SDS. Following lysis, 25 U of Benzonase Nuclease (EMD Chemicals, Gibbstown, NJ) and a 100× phosphatase inhibitor cocktail were added to the lysate. To prepare the samples for analysis, 4× Laemmli sample buffer (Bio-Rad, #1610747), consisting of 950 μL of sample buffer and 50 μL β-mercaptoethanol (Sigma, #M7522), was added. The mixture was then heated at 95 °C for 10 min. Subsequently, 15 μL of the protein sample was loaded onto an SDS-PAGE gel using a Protean TGX precast gradient gel (4%−20%) (Bio-Rad, #17000546) and transferred onto a nitrocellulose membrane (Bio-Rad, #1620115) using a semi-dry transfer system (Bio-Rad). The membranes were blocked for 1 h with 5% BSA, followed by incubation overnight at 4 °C with primary antibodies on a rocker. The primary antibodies used were AMBN (Santa Cruz #sc-271012, 1:500), SP6 (Atlas #HPA024516, 1:1 000), DLX3 (Abnova #H00001747-M09, 1:500), DSPP (Santa Cruz #7363-2, 1:500), H3 (Abcam #Ab1791, 1:1 000), and β-Actin (Cell Signaling #13E5, 1:10 000) all prepared in 5% BSA. The following day, the membranes were washed three times with 1× TBST at 10-min intervals. Afterward, they were incubated for 1 h at room temperature with an anti-rabbit IgG HRP-conjugated secondary antibody (Bio-Rad, #1721019) (1:10 000) and an anti-mouse IgG HRP-conjugated secondary antibody prepared in 5% milk. After incubation, the membranes were rewashed with 1× TBST (three times, 10-min intervals) and developed using the Immobilon Luminol reagent assay (EMP Millipore). Finally, protein bands were visualized using a Bio-Rad ChemiDoc Imager.
RNA extraction and QRT-PCR analysisRNA was isolated from the cells utilizing Trizol reagent (Life Technologies) in accordance with the manufacturer’s guidelines. RNA samples underwent treatment with Turbo DNase (Thermo Fisher Scientific) to eliminate genomic DNA contamination and were subsequently quantified utilizing the Nanodrop ND-1000 spectrophotometer (Thermo Fisher Scientific). For cDNA synthesis, 1 µg of RNA was reverse transcribed using the iScript™ cDNA Synthesis Kit (Bio-Rad) or the Applied Biosystems™ High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Quantitative real-time PCR (qPCR) was performed on 10 ng of cDNA per reaction using SYBR Green (Applied Biosystems) and the 7300 Real-Time PCR System (Applied Biosystems). The conditions for the PCR were established as follows: an initial incubation at 50 °C for 2 min, followed by a denaturation step at 95 °C for 10 min. This was succeeded by 40 amplification cycles, each consisting of a denaturation phase at 95 °C for 15 s and an annealing/extension phase at 60 °C for 1 min. All quantitative PCR (qPCR) reactions were conducted in triplicate, utilizing β-actin (Forward: TCCCTGGAGAAGAGCTACG, Reverse: GTAGTTTCGTGGATGCCACA) as the endogenous control. The comparative ΔCt (threshold cycle) method was employed to evaluate relative gene expression, and the primer sequences used for specific markers of odontoblast differentiation included NESTIN (Forward: GAAACAGCCATAGAGGGCAA, Reverse: TGGTTTTCCAGAGTCTTCAGTGA) and DSPP (Forward: TGACAGCAATGATGAGAGTG, Reverse: CACTGGTTGAGTGGTTACTG).
RNA sequencing library preparation and analysisTotal RNA was extracted from six iAM organoid samples (two biological replicates per condition). RNA quality and concentration were assessed using the Agilent High Sensitivity D5000 ScreenTape® system (cat# 5067-5592, VWR cat# 76645-084) and the Qubit RNA HS Assay Kit (Thermo Fisher Scientific, cat# Q32852), following the manufacturer’s instructions. High-quality RNA was used for library construction with the Illumina Stranded mRNA Prep, Ligation kit (16 samples, cat# 20040532), according to the manufacturer’s protocol. Library size distribution was evaluated with the D5000 ScreenTape, indexed with Illumina adapters, and purified using AMPure XP beads (Beckman Coulter, cat# A63880). Sequencing was performed on the Illumina NextSeq 2000 platform using a P2, 200-cycle sequencing kit (cat# 20100986), generating paired-end 100 bp reads. Reads were aligned to the human reference genome (Ensembl GRCh38) with HISAT2 (v2.2.1), and gene-level counts were generated using FeatureCounts with GENCODE GRCh38 annotations. Principal component analysis was performed with DESeq in R, while differential expression analysis was carried out using DESeq2. Gene Ontology enrichment was assessed using the TopGO R package and the DAVID 6.8 online tool. RNA-seq data have been deposited in the NCBI GEO under accession number (GSE307437).
Mouse single-cell datasetsTo cross-validate findings from the human dataset, we analyzed two independently published mouse scRNA-seq datasets.28,53 The first dataset, cited in Figs. S1 and S5, is a curated integration of mouse dental tissues from multiple studies, available as a loom file from Mendeley Data54 (https://data.mendeley.com/datasets/2kskdknngb/1). Loom files were converted into AnnData objects and used to generate heatmaps. The second dataset comprises whole mouse embryos from embryonic day 8 (E8) to birth (postnatal day 0, P0) and is accessible via CELLxGENE (https://cellxgene.cziscience.com/collections/45d5d2c3-bc28-4814-aed6-0bb6f0e11c82). For this dataset, the dental epithelium cluster was extracted from the whole epithelial dataset and analyzed using the standard Scanpy clustering workflow. Clusters were annotated based on known marker genes.
Pseudotime trajectory visualizationPseudotime expression trajectories for ameloblast markers were generated using diffusion pseudotime values from the dental epithelium subset. For each selected marker gene (Dlx3, Pitx2, Vwde, Dspp, Ambn, Amelx, Mmp20, Enam, Amtn, Klk4, Tuft1, Wdr72, Odam), expression values were ordered by pseudotime and smoothed using a Gaussian kernel (σ = 35) via scipy.ndimage.gaussian_filter1d. Cluster intervals corresponding to known ameloblast states (OE, DE, pAM, eAM, sAM, mAM) were overlaid as color-coded bars along the x-axis. Plots were created in Matplotlib (v3.8.0) with standardized styling (Fig. S3d). A further-smoothed version of these trajectories, generated by downsampling and applying a light Savitzky–Golay filter, including those for Dspp, Enam, and Wdr72, was incorporated into the schematic summary (Fig. 7) to illustrate transcriptional dynamics across ameloblast maturation.
Bulk-to-single-cell correlation analysisTo identify single cells most transcriptionally similar to our organoid bulk RNA-seq profiles, we computed Pearson correlation coefficients between each cell in the mouse dataset and reference bulk expression vectors for each group (C3-DLL4 Treated, Control) using scipy.stats.pearsonr (Fig. S3c). Analyzes were restricted to a shared set of genes between the bulk and single-cell datasets. For each group, the top 15% of cells (≥85th percentile correlation) were defined as the highest-matching subset. These cells were visualized on precomputed UMAP embeddings, with all other cells shown in gray to highlight group-specific similarity patterns. Figures were generated in Matplotlib with consistent sizing, color coding, and scaling. This correlation-based mapping enabled the identification of single-cell populations closely matching bulk transcriptional signatures, as presented alongside cluster-level annotations.
Notch activation and inhibition in ameloblast organoid differentiationhiPSCs were differentiated into 2D induced ameloblasts (iAMs) from D0 to Day 16. At Day 16, iAMs were transferred to low-attachment plates to form 3D ieAM organoids in Epicult Plus medium (Stemcell Technologies #06070). Notch activation was induced at Day 24 using the C3-DLL4 Notch activator (50 nmol/L), a trimeric scaffold conjugated to DLL4, or it was incubated with Notch inhibitor, DAPT (10 μmol/L) (SantaCruz Biotech #sc-201315) (Fig. S2a), for 24 h. On Day 25, the media was replaced, and the ieAM organoids continued maturing into isAMs by Day 31 (Fig. S2a). Likewise, DLX3 knockout clones (Clone 10 and Clone 13) were differentiated following the same protocol.
Immunostaining and confocal imaging of iAM organoidsFor immunostaining, the organoids were fixed in 4% paraformaldehyde (PFA), then immersed in 1× PBS for 3 × 5-min washes and then immersed in 0.5% TritonX 100 at RT for 10 min to facilitate permeabilization. Later, Organoids were blocked in solution (1× PBS containing 10% BSA, 5% normal goat serum, and 0.1% Triton-X) for 2 h. All incubations were done in tubes on a nutator. They were then suspended in antibody dilution buffer (1× PBS containing 10% BSA, 5% normal goat serum, and 0.2% Triton-X) and primary antibodies diluted at concentrations recommended by the manufacturer and incubated overnight at 4 °C. On the second day, cells were washed 3× for 6 min with 1× PBS, resuspended in antibody dilution buffer containing secondary antibodies at 1:200 and DAPI (1:50), and then incubated overnight at 4 °C. On the third day, organoids were washed 3× for 6 min in 1× PBS and mounted in Vectashield Antifade Mounting Media on a glass concavity microscope slide, one to three organoids per well. Organoids were then imaged using a Leica (DMi8) SP8 LIGHTNING confocal microscope (25× and 40× objectives) equipped with HyD and PMT spectral detectors and Leica LASX acquisition software [version 3.5.5IR], or a High Resolution Widefield Nikon ECLIPSE Ti Fluorescent microscope and Nikon Ti2 confocal microscope with Nikon NIS-Elements software.
Quantification of immunofluorescence signalsImmunofluorescence signals for the proteins of interest (ENAM, AMELX, and MMP20) were quantified as follows. First, raw images were processed using Fiji (ImageJ v2.3.091/92) to split each image stack into individual TIFF files corresponding to DAPI (4′,6-diamidino-2-phenylindole) and the target protein. To normalize quantification across each organoid, nuclei were segmented using the StarDist3D pipeline on ZEISS arivis Cloud (Apeer) and then counted with ImageJ’s 3D Object Counter, ensuring that threshold parameters remained consistent across images acquired under similar conditions.
The fluorescent signals for the proteins of interest were quantified in three dimensions by segmenting puncta with the ImageJ 3D Object Counter. The ImageJ measure function was then used to determine both the mean fluorescence intensity and the integrated intensity for each punctum. The overall mean signal for each organoid was normalized to the nuclei count. Finally, all results were compiled into a spreadsheet and analyzed with GraphPad Prism to assess the statistical significance between groups and conditions.
Kidney capsule transplantationAll animal procedures were performed in compliance with relevant ethical regulations and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Washington, Seattle. Adult male NOD-SCID mice (RRID: IMSR_JAX:001303), 8 weeks of age, were used for all experiments. Mice were housed under specific pathogen-free conditions with ad libitum access to standard chow and water. ismAM organoids were differentiated for 25 days following the protocol described above, with C3-DLL4 treatment throughout. On the day of transplantation, organoids in suspension were transferred into beveled, kinked PE50 tubing (BD Intramedic, #427517). Adult male NOD-SCID mice (n = 6) were anesthetized in an induction chamber with isoflurane in oxygen, then maintained under 2% isoflurane via a nose cone. Body temperature was maintained at 37 °C using a heating pad for the duration of the procedure. The dorsal flank was shaved and sterilized with betadine, followed by alcohol wipes, before making a 1–2 cm incision. The kidney was gently externalized with a cotton swab, and the capsule was nicked near the caudal end using a 22-gauge needle. The beveled PE50 tubing was inserted beneath the capsule, and the organoid suspension was delivered under the control of a Hamilton syringe. The capsule opening was sealed with a cotton swab to promote clotting and retain the graft. The kidney was then returned to the abdominal cavity, the peritoneum was closed with absorbable sutures, and the skin was closed with surgical staples. Following surgery, mice were placed in a heated recovery cage until fully awake, then returned to their home cages and transported back to the vivarium. All procedures were performed in compliance with ethical regulations under IACUC protocol #4152-01. At 21 days post transplantation, mice were euthanized via CO₂ inhalation. Kidneys with intact grafts were harvested, fixed in 4% paraformaldehyde at 4 °C for 1 h, transferred to 15% sucrose for 2 h, and then to 30% sucrose overnight at 4 °C. Kidneys were bisected and embedded in embedding cryo-molds (Sakura, #25608-916) with Tissue-Tek O.C.T. compound (Sakura, #4583). The embedded tissue was snap-frozen by immersion in 2-methylbutane (EMD, #MX0760-1) chilled in a liquid nitrogen slurry to −80 °C, then stored at −80 °C until sectioning. Cryosections (15 µm) were cut on a pre-chilled cryostat (−20 °C) onto Superfrost Plus microscope slides (Fisherbrand, #12-550-15) and stored at −80 °C until further analysis.
Micro-computed tomography (microCT)As a proof of concept, we used high-resolution microCT imaging to identify calcified tissue within a sample organoid implanted kidney. We adapted methods from a previous study33 in order to enable full 3D reconstruction of the kidney without manual edge detection while not desiccating the specimen. Briefly, we 3D printed an imaging platform that press fit within a 15 ml Falcon tube, filled the distal 13 mL of the tube with 30% sucrose-soaked gauze, inserted the platform into the proximal end of the tube, placed the kidney on the platform, and sealed the tube. High-resolution microCT images were then obtained for the entire specimen (Scanco vivaCT 40; 10.5 μm voxel size, 45 kVp, 177 μA). The raw image data were preprocessed using a Gaussian Filter algorithm to remove image noise (Sigma = 1.2, Support = 2.0), followed by segmentation of calcified material within the specimen using standard image thresholding techniques and, finally, manual comparison of the grayscale and binary images to confirm that threshold values maintain morphologic fidelity (201 mg HA/cm3).55,56 For each 2D image, the surface of the kidney was identified through an automated secondary thresholding procedure to delineate soft tissue. The resulting 3D image depicts the kidney (rose color) and calcified tissue(blue).
Histological analysisHematoxylin and eosin (H&E) stainingFor hematoxylin and eosin (H&E) staining, the frozen fixed tissue sections were air-dried at room temperature for 10 min and subsequently fixed in 4% paraformaldehyde (PFA) for 10 min. Slides were rinsed in PBS and incubated with Mayer’s hematoxylin for 5 min, followed by a 5 min wash in running tap water and bluing in 0.2% ammonia water for 1 min. Slides were then counterstained with eosin Y for 2 min, dehydrated through a graded ethanol series (70%, 95%, 100%) and cleared in xylene. Coverslips were mounted using a resinous mounting medium (Permount, Fisher Scientific), and images were acquired using Nikon NSPARC brightfield microscope.
Trichrome stainingFor modified Masson’s Trichrome staining, the frozen fixed tissue sections were air-dried and incubated in pre-warmed Bouin’s solution at 56 °C for 1 h. Slides were cooled to room temperature and washed in running tap water for 10 min until the yellow color had cleared completely. Sections were stained with Weigert’s iron hematoxylin for 10 min, rinsed in tap water, and incubated in Biebrich scarlet-acid fuchsin solution for 10 min. Differentiation was performed in phosphomolybdic/phosphotungstic acid solution for 15 min without rinsing. Slides were then transferred directly to aniline blue solution for 10 min, rinsed in 1% acetic acid for 1 min, and dehydrated through ethanol and xylene. Coverslips were mounted using either resinous medium for permanent brightfield imaging or aqueous mounting medium (e.g., VECTASHIELD) for subsequent immunofluorescence analysis. Representative images were captured using Nikon NSPARC brightfield microscope.
Von Kossa and Alizarin Red S stainingFor Von Kossa staining, frozen sections fixed in 4% paraformaldehyde (EMS, #15710) were incubated in 5% silver nitrate solution (Sigma-Aldrich, #209139) under ultraviolet light for 1 h 30 min. Sections were rinsed several times in deionized distilled water (5 min each rinse), then incubated in 5% sodium thiosulfate solution (Sigma-Aldrich, #217263) for 5 min to remove unreacted silver.
For calcium detection, sections were stained in 1% Alizarin Red S solution (pH4.2) (Sigma-Aldrich, #A5533) for 30 min in the dark, followed by thorough rinsing in deionized distilled water (5 min each rinse). Sections were counterstained with either nuclear fast red (EMS, #26078-05) or 0.2% aqueous Fast Green FCF solution (EMS, #26053-02) for 30 s, then briefly rinsed in deionized distilled water (5 min each rinse). Slides were mounted with Permount Mounting (#SP15100-EA) and stored at room temperature until imaging.
Graphics and illustrationsThe illustrations in the graphical abstract and in Figures were created with BioRender.com.
Statistical analysisAll quantifications show the mean, and error bars are ± SEM. Ordinary one-way ANOVA was used for multiple comparisons. A two-tailed, unpaired t-test was used for comparing groups of two using GraphPad Prism. P-values < 0.05, 0.01, 0.001, 0.000 1 are indicated with *, **, *** and ****, respectively. Software used and methods for analysis and quantification of each data in this manuscript are described in the method section.
Comments (0)