In this study, we conducted a comprehensive analysis of the pathogenicity of SFTPC gene mutations in pediatric patients at our center, integrating clinical and genetic data to provide valuable insights for researchers and clinicians. Furthermore, through a systematic review of previously reported cases in the Human Genome Variation Society (HGVS) and Human Gene Mutation Database (HGMD), this work offers a broader perspective on genotype-phenotype correlations. The study aims to deepen the understanding of this rare lung disorder and inform the development of future diagnostic and therapeutic strategies.
Surfactant protein C (SP-C) deficiency is a rare autosomal dominant genetic disorder characterized by progressive respiratory insufficiency and interstitial lung diseases (ILDs), with highly variable clinical courses [9]. The prevailing pathophysiological hypothesis posits that intracellular accumulation of structurally defective SP-C protein disrupts surfactant homeostasis and alveolar epithelial integrity [10]. Pulmonary surfactant metabolism disorder 2 (SMDP2, OMIM: 610913) is caused by heterozygous mutations in the SFTPC gene, which encodes SP-C. Since the first description by Nogee et al. in 20016, two decades of research on SFTPC-associated ILDs have elapsed, driving continuous advances in our understanding of genotype-phenotype relationships and clinical management strategies.
In the HGMD, mutations are classified into five categories. Disease-causing mutations (DM) are defined as pathological mutations that have been reported in at least one literature to cause disease. Suspected disease-causing mutations (DM? ) refer to mutations reported as pathogenic in literature, but with authorial doubts or subsequent evidence indicating questionable damaging effects. Disease-associated functional polymorphisms (DFP) denote mutations with a population allele frequency > 1% that are significantly associated with a specific disease or phenotype and have been confirmed to exert functional effects (e.g., impacting gene structure, expression, or product function). Disease-associated polymorphisms (DP) represent mutations with a population allele frequency > 1% that exhibit significant association with a disease or phenotype, yet lack proven functional impact. In vivo/in vitro functional polymorphisms (FP) refer to mutations reported in literature to affect structure, function, gene expression, or gene products, but which have not yet been linked to any disease. Meanwhile, we employ the systematic gene mutation nomenclature established by the HGVS, which is currently the universally accepted naming convention in academia. For detailed guidelines on gene mutation nomenclature, please refer to the website http://www.HGVS.org/varnomen8.
To date, 92 unique mutation sites have been identified in the SFTPC gene, with the majority localized to exons 3, 4 and 5, most frequently within the exon 4 region. Notably, while three promoter region mutations are categorized as DFP in HGMD, they demonstrate substantial translational potential for basic research on surfactant biology (Supplementary Figs. 1 A, 1B, 1D).
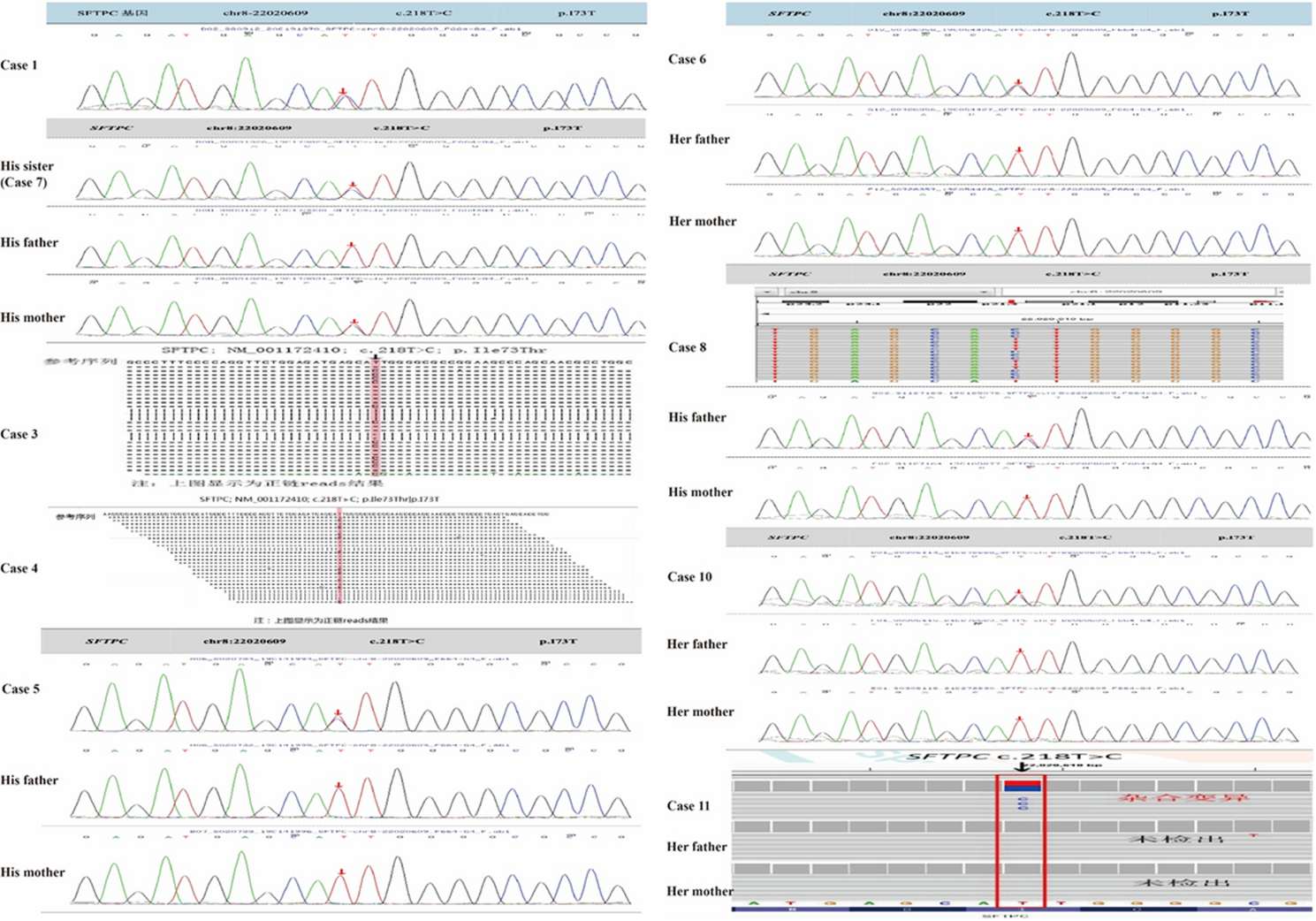
In our cohort, 9 cases carried mutations in exon 3, 1 case in exon 2, and 1 case in exon 5. The c.218T > C (p.Ile73Thr) variant was the most prevalent, accounting for approximately 30% of all mutations consistent with prior reports [11]– [12] identifying this canonical hotspot mutation. Of the 11 cases, 9 harbored this specific locus alteration. Additionally, we observed that certain SFTPC mutations were associated with non-ILD phenotypes, including bronchial atresia [13]bronchiectasis [14]and pulmonary fibrosis [15]– [16]. Notably, the newly identified splicing variant (c.612 + 1G > T) at our center presented uniquely with pulmonary atelectasis (PA) as the primary clinical manifestation, whereas other mutations predominantly caused ILDs. These variants’ distinct genotype-phenotype correlations warrant further mechanistic investigation (Supplementary Fig. 2).
Regarding genetic inheritance, Nogee et al. first reported familial clustering of ILDs associated with SFTPC mutations [17]. Subsequently, Thomas et al. described a large pedigree with heterozygous SFTPC variants, where affected individuals showed phenotypic variability in ILD severity despite shared SP-C deficiency. Notably, two asymptomatic obligate carriers were identified in this family, indicating reduced penetrance of the genetic defect [18]. Subsequent studies have further shown that viral infections may influence the penetrance of SFTPC mutations, modulating clinical disease expression [19]– [20]. In our cohort, sporadic cases constituted approximately 50% of patients, consistent with the reported balance between familial and sporadic cases in prior literature (Supplementary Table 1). The almost equal distribution of sporadic and familial inheritance patterns of SFTPC mutations in our cohort underscores the ongoing need for comprehensive family genetic screening, even in confirmed cases.
As previously documented, approximately 72.9% of SFTPC mutation sites are localized within the BRICHOS domain [21]. Compared to mutations in the mature SP-C or linker regions, these BRICHOS-domain mutations are characterized by significantly earlier onset ages and narrower age ranges. Clinically, BRICHOS-domain mutations are associated with higher case fatality rates, often presenting as respiratory distress syndrome (RDS), early-onset dyspnea and severe disease requiring prolonged mechanical ventilation [22,23,24,25,26]. In contrast, non-BRICHOS mutations more commonly manifest with chronic phenotypes, including persistent cough, exertional dyspnea, failure to thrive, and recurrent respiratory tract infections [27]. Notably, our longitudinal analysis found no significant temporal variation in the frequency of BRICHOS versus non-BRICHOS domain mutations. The severity of SFTPC-related ILDs remains difficult to systematically quantify, owing to multifactorial influences from genetic background, environmental triggers, and individual host responses (Supplementary Figs. 1 C and 2).
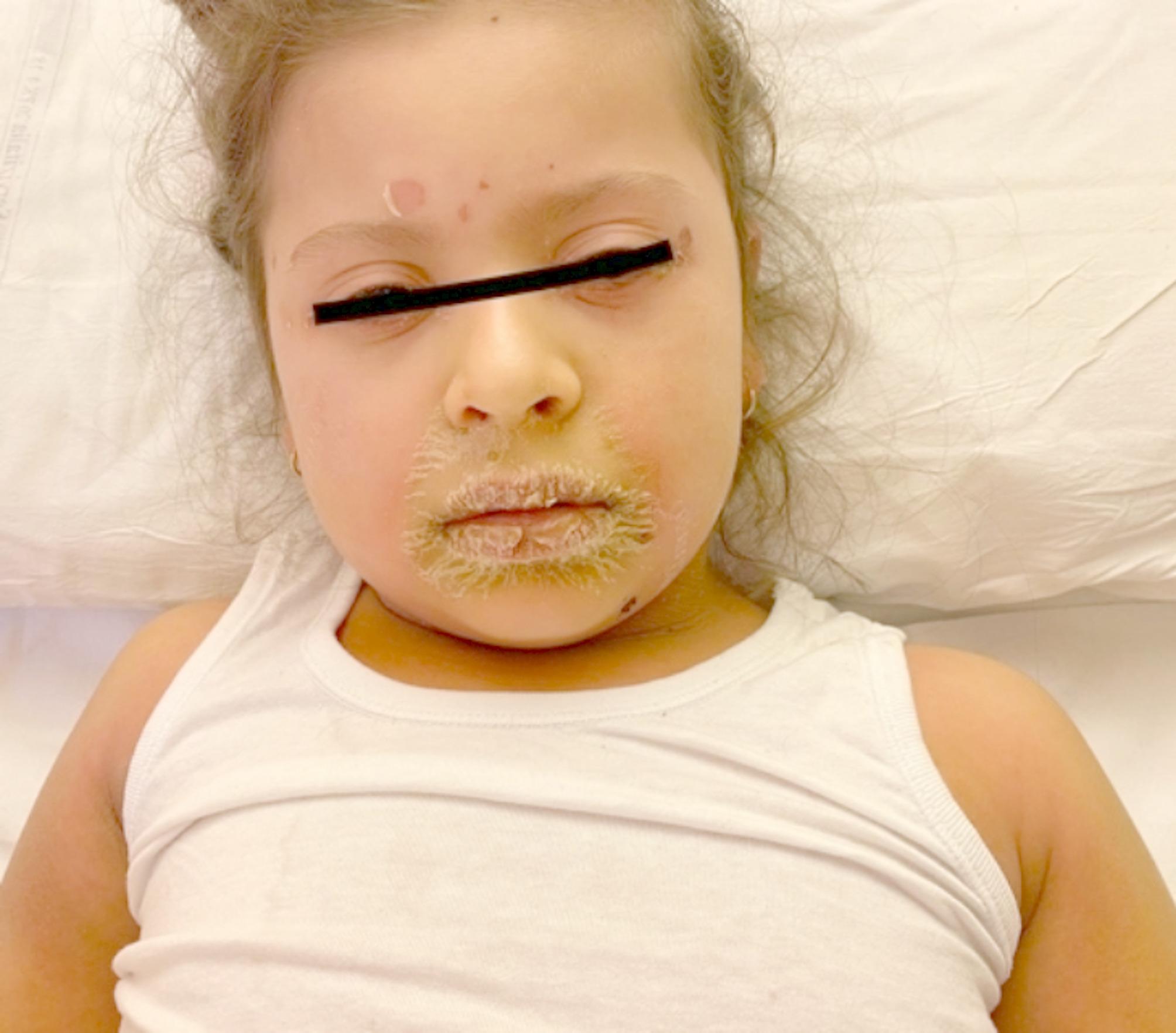
Clinical manifestations of SFTPC-related lung diseases display remarkable heterogeneity, tightly linked to age at onset: neonatal respiratory distress or failure [28]– [29]childhood chronic cough [30]recurrent pulmonary infections, wheezing, exertional dyspnea [31]and in adulthood, classic restrictive ventilatory dysfunction features such as reduced activity tolerance, weight loss, chronic hypoxia, and digital clubbing [32]. However, these symptoms are nonspecific and demand careful discrimination from other ILDs. The clinical profiles of our cohort mirror prior observations, lacking disease-specific biomarkers. This highlights the necessity of collaborative assessment involving clinicians, radiologists, pulmonologists, and geneticists.
Over the past two decades, bronchoalveolar lavage (BAL) has established itself as a cornerstone in the management of ILDs. The 2002 ATS/ERS International Consensus Classification of Idiopathic Interstitial Pneumonia (IIP) underscored the utility of BAL in differentiating etiologies of ILDs [33]. Notably, BAL serves a dual role as both a diagnostic modality and first-line therapeutic intervention for pulmonary alveolar proteinosis [34]– [35]. In our institutional experience, a patient harboring an SFTPC splicing variant (c.612 + 1G > T) with PA exhibited marked symptomatic improvement following BAL. These observations further validate the escalating significance of BAL in the comprehensive management of SFTPC-associated ILDs, demonstrating its capacity to bridge diagnostic ambiguity and therapeutic intervention.
Chloroquine was first reported for the treatment of pediatric interstitial lung diseases (ILDs) in 1994, with early case series demonstrating promising efficacy [36,37,38,39]. However, to date, only approximately 10% of patients have received hydroxychloroquine (Hch) (Supplementary Table 1), primarily due to concerns over age-appropriate immunosuppressant dosing, substantial side effect profiles, and ethical considerations. These factors pose challenges that complicate clinical decision-making. In our cohort, Cases 1, 7, and the affected mother demonstrated remarkable improvement in both clinical symptoms and lung imaging after initiating Hch treatment. This positive therapeutic response notably enhanced the confidence of Case 11’s parents, leading them to consent to Hch therapy, which has since effectively maintained disease control. Glucocorticoids are well-established for their potent anti-inflammatory and bronchodilatory properties. However, their significant adverse event profiles, especially when administered at high doses, often deter many caregivers from prescribing them, highlighting the need for alternative therapeutic options like Hch.
Azithromycin (AZP) has seen growing clinical application, underpinned by preclinical evidence demonstrating its capacity to mitigate amyloid-like deposition of misfolded SP-C protein, a proposed key pathogenic intermediate in SFTPC-related diseases [40]. In accordance with this mechanistic rationale, all pediatric patients at our center received AZP as an integral component of their management protocol. Although lung transplantation is considered a potential definitive treatment for advanced ILDs, its long-term efficacy remains uncertain. This ambiguity stems from limited case numbers and the paucity of systematic longitudinal follow-up data [41,42,43].
This study is subject to several limitations. Firstly, despite our efforts to comprehensively collate all published SFTPC-related cases worldwide, the extreme rarity of the disorder posed challenges. Excluding the 11 cases from our center, a significant portion of the data was sourced from literature reviews. The restricted access to primary clinical data led to incomplete phenotypic characterizations and follow-up information for certain reported cases, which may have introduced selection bias into the statistical analyses. Secondly, most existing reports predominantly offer cross-sectional clinical details at the time of diagnosis, yet lack longitudinal follow-up data. This deficiency hinders a thorough assessment of long-term prognosis and disease progression trajectories. These constraints underscore the urgent necessity for international collaborative registries that can aggregate standardized data, thereby enabling comprehensive natural history studies of this rare lung disorder.



Comments (0)