
Remember me

For model construction, we collected, in total, five kinds of datasets in our study: a large-scale proteome, AMPs, non-AMPs, an external validation dataset, and a toxin and non-toxin dataset.
ProteomeNon-redundant canonical and isoform sequences (609,216) were retrieved from UniProt (http://www.uniprot.org/) (downloaded as of October 2022). The protein library composed of these sequences serves two main purposes: pre-training the ProteoGPT model and constructing different types of NRSPD.
(1)Pre-training the ProteoGPT model
Due to computational constraints and model requirements, all sequences were segmented into subsequences with a maximum length of 1,000 AAs to serve as a training corpus.
(2)Constructing different types of NRSPD
A sliding window with a size of L was employed to scan all protein sequences for constructing multiple NRSPDs. To avoid windows with >50% sequence overlap, we set the sliding step size to L/2. In our study, L was set to 8–30. Five NRSPDs were constructed to be the subject for data mining of AMPs: 8–10 AAs, 11–15 AAs, 16–20 AAs, 21–25 AAs and 26–30 AAs, totalling 410,192,277 non-redundant short peptides.
AMPsAMP data were primarily sourced from four public AMP datasets: APD3 (ref. 37), DBAASP38, DRAMP39 and CAMP40, which cover most AMP sequences from various origins (downloaded as of November 2022). AMPs exhibiting antibacterial, antiviral and antifungal activities, and having lengths under 50 AAs, were deduplicated and consolidated into an extensive dataset, resulting in 16,062 non-redundant AMPs used for subsequent analysis.
Non-AMPsWe compiled the non-AMP sequences from UniProt, applying the ‘subcellular location’ filter to cytoplasm. Any entries containing keywords such as antimicrobial, antibiotic, antiviral, antifungal, effector or excreted in their functional annotations were excluded (downloaded as of November 2022). We refined the dataset to include only sequences not exceeding 50 AAs in length and removed any duplicates. Concurrently, sequences identical to any known AMPs were eliminated from the non-AMP dataset, resulting in a final tally of 16,549 unique non-AMP sequences. The non-AMP sequence dataset we compiled encompasses a wide range of biological sources, including eukaryotes (for example, humans and other plants and animals), prokaryotes (for example, Escherichia coli and Salmonella enterica) and viruses (for example, HIV-1 and hepatitis B virus) (Supplementary Table 5).
External validation datasetThe AMP sequences in the external validation dataset are distinct from those used in the model construction. These sequences were collected from the APD3 (ref. 37), DBAASP38, DRAMP39, CAMPR4 (ref. 48), LAMP2 (ref. 49) and BaAMPs50 databases. The selection criteria included sequences no longer than 50 AAs and those with antifungal activity stronger than antibacterial activity. In addition, 370 non-AMP sequences (≤50 AAs, without overlap with non-AMPs) were derived from ref. 25. To ensure the independence of the external dataset, any sequences that were part of the model training, validation or testing sets were excluded.
Toxin and non-toxin datasetWe collected 1,932 toxic peptide sequences that were experimentally validated and 1,932 non-toxic peptide sequences with a range of 10–50 residues from ToxIBTL29 (https://server.wei-group.net/ToxIBTL/) (downloaded as of April 2023).
In this study, we collected peptide sequence data generated by five unconstrained generation models (HydrAMP31, Basic31, PepCVAE32, AMP-GAN33, AMP-LM34) to compare their performance in AMP generation with AMPGenix. The following quality control and preprocessing steps were applied to all datasets collected from these models:
Prefix alignmentThe first AA of each sequence was restricted to one of the 10 selected residues from the set G, K, F, R, A, L, I, V, S and W.
Quantity alignmentA maximum of 800 sequences per starting AA residue was retained, resulting in a total of 7,000–8,000 sequences for each model.
Length alignmentSequence lengths were constrained to the range of 5–35 AAs to ensure consistency with the length range of sequences generated by AMPGenix.
After filtering, the final number of valid sequences for each model was as follows: HydrAMP, 8,000; Basic, 7,810; PepCVAE, 7,489; AMP-GAN, 7,906; and AMP-LM, 7,405.
Model buildingWe built, in total, one pre-trained and three transfer LLMs with different functionalities.
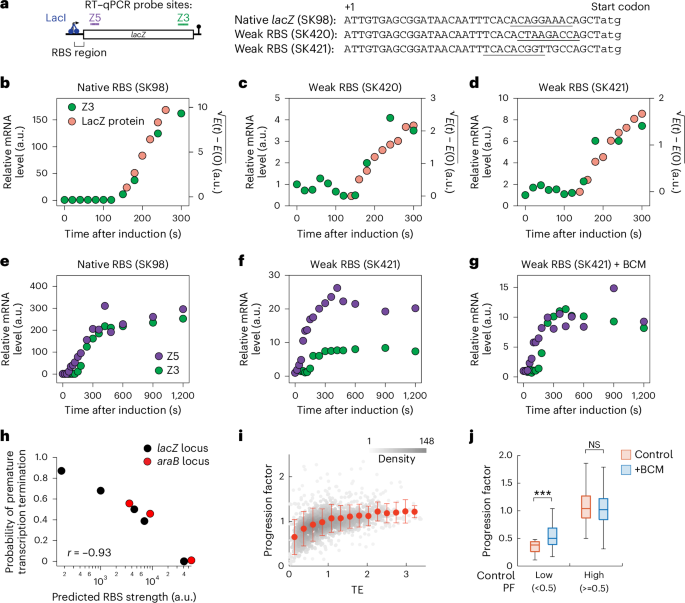
Training dataset, validation dataset and test datasetThe datasets employed for the training of AMPSorter and BioToxiPept were randomly partitioned into training, validation and testing sets at a 6:2:2 ratio (Fig. 2a). These datasets were mutually exclusive. The training set was utilized for model development, the validation set for tuning hyperparameters, and the testing set for assessing the model’s performance. To ensure robust evaluation, a benchmarking set was constructed by applying CD-HIT (v.4.8.1) for sequence filtering. First, sequences with over 90% similarity were removed from the training and validation sets. Then, CD-HIT-2D was used to exclude sequences from the test set that shared >70% identity with any sequence filtered in the first step. In addition, sequences containing UAAs were removed. This resulted in a stringent benchmarking set of 725 AMPs and 1,071 non-AMPs, providing a reliable basis for comparing model performance against existing AMP classification algorithms including AMPlify22, Macrel23, iAMP Pred24, AMP Scanner (v.2)25, Bert-Protein26, AMPir27 and AmPEP28.
Data encoding and decodingAll sequences were encoded and decoded using GPT-2 tokenizer. The tokenizer encoded sequences, converting them into the tokenizer form for model input, and decoded them, transforming model outputs back into AAs. Sequence beginnings/ends were marked with the ‘<|endoftext|>’ label. During the pre-training and fine-tuning of the model, the datasets were divided into multiple pieces to optimize memory usage and balance the training load. This approach is particularly useful when dealing with large datasets, as it helps improve training efficiency and prevent memory overflow.
Model structureThe most fundamental model is ProteoGPT based on GPT-2 architecture. The model structure of AMPGenix is identical to that of ProteoGPT. AMPSorter and BioToxiPept were created by adding an additional classification module at the end of ProteoGPT to reduce the dimension to 2. Comprehensive model structure details can be found in the Supplementary Text.
Model evaluationA cross-entropy loss function was used to train AMPSorter and BioToxiPept.
$$\beginL=-\mathop\limits_^_\log \left(\;p(\;_)\right)\end$$
(1)
where:
N is the number of samples;
yi is the true label of sample i;
p(yi|x) is the predicted probability of sample i by the model, given x.
Accuracy, specificity, sensitivity, precision, F1 score, and MCC, receiver operating characteristic (ROC) curve and PR curve were measured to assess the classification models as follows (TP, true positive; TN, true negative; FP, false positive; FN, false negative; FPR, false positive rate):
$$\begin}=\frac}+}}}+}+}+}}\end$$
(2)
$$\begin}=\frac}}}+}}\end$$
(3)
$$\begin}=}=\frac}}}+}}\end$$
(4)
$$\begin}=\frac}}}+}}\end$$
(5)
$$\begin}=\frac}}}+}}\end$$
(6)
$$\begin}=\frac}}}+}}\end$$
(7)
$$\begin} }=\frac\times }+}}\end$$
(8)
$$\begin}=\frac}\times }-}\times }}}+}\right)\left(}+}\right)\left(}+}\right)\left(}+}\right)}}\end$$
(9)
The ‘auc’ function in the scikit-learn package (v.1.2.2) was used to compute the AUC and AUPRC values.
Uniqueness (proportion of unique generated sequences to the total number), diversity (average pairwise cosine distance between generated sequences), novelty (average cosine distance between generated sequences and known AMPs) and FCD (Fréchet distance measuring the distributional similarity between generated sequences and known AMPs) were used to evaluate the quality and diversity of sequences generated by generative models.
$$\begin}=\frac}}}}}\end$$
(10)
$$\begin}=\frac\mathop\limits_}_}}(_,_)\end$$
(11)
where N is the total number of sequences, and Xi, Xj are the feature vectors for sequences i and j.
$$\begin}=\frac\mathop\limits_^\mathop\limits_^}_}}(_,_)\end$$
(12)
where N is the total number of sequences, M is the number of known AMPs, and Xi, Xj are the feature vectors for the generated and known AMPs, respectively.
$$\begin}=_-_}^+}(_+_-2\sqrt__})\end$$
(13)
where μ1 and Σ1 are the mean and covariance of the generated sequences, and μ2 and Σ2 are the mean and covariance of the known AMPs, respectively.
QSAR analysisThe introduction of the QSAR model has specific background and purpose. To compare data mining and text generation strategies under the same threshold, we incorporated the QSAR model, which offers several advantages:
(1)Clear threshold setting: The QSAR model can provide a clear quantitative threshold, enabling the selection of an appropriate number of peptides for further validation.
(2)Consistent peptide types: The QSAR model, through quantitative structure–activity relationship predictions, predicts the antimicrobial activity of cationic AMPs and ensures that the two groups of peptides being compared are consistent in type, avoiding potential biases from differing peptide types. By introducing QSAR as a third-party tool, we can ensure the selection of consistent types of peptide from the candidate pools, thereby improving the comparability of both strategies in terms of development efficiency and antibacterial efficacy.
Each sequence was scored using the method described in refs. 35,36, which uses peptide charged residues and hydrophobic residues to create a relative score for the propensity of the peptide to present antimicrobial activity:
$$}\; }\left(}\right)=\frac^^}}}$$
(14)
where C represents the net charge, H represents the total hydrophobicity, and MaxScore is the maximum value of CmHn that can be calculated with the given coefficients m = 0.9 and n = 1.1 as described35,36.
Gene function analysisEggNOG mapper (http://eggnog-mapper.embl.de/) was applied to analyse protein-encoding genes with default parameter settings.
Peptides and chemicalsPeptides with purity >95% were custom synthesized by Synthbio Technology. The powder of each peptide was individually dispensed into several tubes of 2 mg per tube and stored in a refrigerator at −80°C. The powder was prepared on the day of the start of each experiment and used at the appropriate concentration. Dimethylsulfoxide (DMSO), cyclophosphamide, DiSC3-(5) and vancomycin were purchased from Mcklin. HEPES, polymyxin B, MTT Cell Proliferation and Cytotoxicity Assay kit were obtained from Solarbio, and PI was obtained from Yuanye. PBS was obtained from Cytiva. Triton X-100 was purchased from Sigma-Aldrich. DMEM/High glucose medium and penicillin–streptomycin mixed solution (Dual antibody) were purchased from Shanghai Zhong Qiao Xin Zhou Biotechnology. d-glucopyranose and potassium chloride (KCl) were obtained from Hushi. Unless otherwise stated, all chemicals were purchased from Solarbio or the China National Pharmaceutical. Details of the reagents are provided in Supplementary Table 6.
Strains and mediaE. coli ATCC25922, S. aureus ATCC25923 and C. albicans ATCC10231 were purchased from the Beijing Microbiological Culture Collection Center (BJMCC). ICU- isolated CRAB QLH-2022-267, CRAB QLH-2022-636, CRAB QLH-2022-637, MRSA QLH-2022-266 and MRSA QLH-2022-718 were provided by the Qilu Hospital Strain Bank, Shandong University. In this study, E. coli ATCC25922, S. aureus ATCC25923, CRAB strains and MRSA strains were cultured in Luria Bertani (LB) broth/agar medium. C. albicans ATCC10231 was cultured in yeast extract peptone dextrose (YPD) broth/agar medium. Mannitol salt agar (MSA), MacConkey agar (MCA), Mueller–Hinton broth (MHB), Mueller–Hinton agar (MHA) and tryptone soybean broth/agar (TSB/TSA) media were used in this study. All media were supplied by Hopebiol. Unless otherwise specified, all strains were incubated at 37 °C.
MiceAll animal experiments were performed according to the ‘Principles of laboratory animal care’ (NIH publication No. 86–23, revised 1985) and approved by the Animal Care and Use Committee of Shandong University (LL20240622). Male BALB/c mice (5-week-old) were purchased from Beijing Vital River Laboratory Animal and kept under a 12 h light/12 h dark cycle, humidity of 50% and temperature of 22 °C in standard specific-pathogen-free (SPF) individually vented cages.
Antimicrobial activity assaysTo screen AMPs for better antimicrobial efficacy, a modified Kirby–Bauer disk diffusion assay was used. E. coli ATCC25922 and S. aureus ATCC25923 were cultured to log phase. MHA plates were used as the base medium. A 0.75% agar solution was prepared, sterilized and cooled to ~55 °C. Then, 100 μl of the bacterial suspension was added to 7 ml of the molten agar, mixed thoroughly and poured onto the surface of the pre-solidified MHA base plate to form a double-layer agar plate. The plates were allowed to solidify at room temperature. Sterile filter paper discs (7 mm in diameter) were placed on the surface of the double-layer plates, and the test compound was added to the centre of each disk. All plates were incubated at 37 °C for 16 h. After incubation, the diameter of the inhibition zones was measured in millimetres and recorded to evaluate the antimicrobial activity of the compounds. All 196 peptides were first selected at a content of 128 μg and further selected at a content of 64 μg (Extended Data Fig. 3). To determine MIC values, 58 selected peptides were dissolved into MHB with an initial concentration of 2,560 μg ml−1, followed by 2-fold serial dilutions. Cells of E. coli ATCC25922 and S. aureus ATCC25923 were separately suspended in the assay medium at a density of 1 × 108 c.f.u.s ml−1. Then, 100 μl of each AMP solution was added to 100 μl of the bacterial suspension in a 96-well plate. Plates were incubated at 37 °C for 16 h. Finally, 20 candidate AMPs were selected using the broth microdilution technique46 in MHB. Next, these AMPs were dissolved into MHB with an initial concentration of 1,024 μg ml−1, followed by 2-fold serial dilutions. E. coli ATCC25922, S. aureus ATCC25923, CRAB QLH-2022-267, CRAB QLH-2022-636, CRAB QLH-2022-637, MRSA QLH-2022-266 and MRSA QLH-2022-718 were cultured in LB at 37 °C and 120 r.p.m. to log phase. C. albicans ATCC10231 were cultured in YPD at 30 °C and 120 r.p.m. to log phase. Cells were separately suspended in the assay medium at a density of 105 c.f.u.s ml−1. Then, 100 μl of each AMP solution was added to 100 μl of the bacterial suspension in a 96-well plate. Plates were incubated at 37 °C for 16 h. The optical density (OD) values of 625 nm were measured using a TECAN Spark microplate reader following a previous report47.
Haemolysis effect of candidate AMPsFreshly collected sheep red blood cells bought from Solarbio were first washed with PBS until the upper phase was clear after centrifugation (491 × g) and allocated onto 96-well flat-bottom plates. Each AMP was diluted and added to the well at a final concentration corresponding to its maximum MIC values against E. coli ATCC25922 and S. aureus ATCC25923 under high inoculum condition of 108 c.f.u.s ml−1. After 1 h at 37 °C, cells were centrifuged at 2,996 × g for 10 min. PBS and Triton X-100 were used as negative and positive controls, respectively. The supernatant was removed and OD450 was measured. All experiments were performed with three independent replicates. The haemolysis rate was calculated using the formula:
$$}\; }\left( \% \right)=\frac}(})}}(})}}}-100)}}(})}\times 100 \%$$
(15)
Cytotoxicity against mammalian cellsCytotoxicity of candidate AMPs was determined using the MTT Cell Proliferation and Cytotoxicity Assay kit46. HEK293 cells (FUNHENG BIOLOGY) were inoculated in 96-well flat-bottom plates at 5,000 cells per well in cell culture medium. After 24 h incubation at 37 °C with 5% CO2 in the atmosphere, the medium was replaced with fresh medium, and AMPs (final concentration: 100 μg ml−1) were added, followed by 48 h incubation. Cell viability was monitored by adding MTT solution and measuring OD490 after 4 h. Zero wells and control wells were set during this experimental process. All experiments were performed with three independent replicates. Cell survival rate was calculated using the formula:
$$}\; }\; }\left( \% \right)=\frac(\rm)}}\rm(\rm)}(\rm)}}\rm(\rm)}\times 100 \%$$
(16)
Modelling and treatment of neutropenic thigh infections in miceMice were injected intraperitoneally with cyclophosphamide 4 d and 1 d before bacterial administration, at concentrations of 150 mg kg−1 and 100 mg kg−1, respectively, to induce neutropenia35,47. MRSA QLH-2022-718 and CRAB QLH-2022-637 were suspended separately in sterile PBS, adjusted to a concentration of ~106 c.f.u.s per infection site and injected into the right thighs of mice in the corresponding experimental groups. Then, AMPs (10-fold MIC, 100 μl; Nmice = 7 for most AMP groups; Nmice = 6 for m_AMP76 group with MRSA infection; Nmice = 9 for g_AMP33 group with CRAB infection; Nmice = 8 for m_AMP76 group with CRAB infection), sterile water (Nmice = 8 with CRAB infection, Nmice = 7 with MRSA infection), polymyxin B (20,000–25,000 U kg−1 day−1, Nmice = 10) or vancomycin (40 mg kg−1, Nmice = 8) were given intraperitoneally at 1, 3, 5 and 7 h after infection. At 24 h after infection, mice were euthanized and thigh wound tissue was collected, weighed, homogenized and serially diluted in sterile PBS. C.f.u.s of MRSA QLH-2022-718 and CRAB QLH-2022-637 were calculated for each thigh wound tissue by diluting the thigh wound homogenate (0.25 g of thigh tissue in 10 ml sterile PBS), inoculating it on MSA plates and MCA plates, respectively, and counting the colonies.
Histologic changes in the kidney and colon of miceTissue samples were fixed in 4% paraformaldehyde for at least 24 h, dehydrated in a graded ethanol series, cleared in xylene and embedded in paraffin. Sections (4 μm) were cut, mounted on glass slides and dried at 60 °C. Slides were dewaxed, rehydrated, stained with haematoxylin (3–5 min), differentiated, blued, counterstained with eosin (15 s), dehydrated, cleared and mounted with neutral balsam. Images were acquired using a light microscope.
16S rRNA sequencing and analysisThe DNA of mouse caecum and colon contents were extracted using the EasyPure Stool Genomic DNA kit (EE301-01, TransGene) and sequenced on the Illumina Nova 6000 platform. Shannon index was calculated using QIIME2 (ref. 51). Euclidean distance matrix was computed with the ‘dist’ function and plotted using the ‘hclust’ function (base R package). Principal coordinates analysis (PCoA) and permutational multivariate analysis of variance (PERMANOVA) were conducted using adonis2 (vegan R package).
Resistance to proteolytic degradation assaysAMPs were incubated in fetal bovine serum (FBS) to evaluate resistance to enzymatic degradation45. Peptides were exposed to an aqueous solution of 25% FBS at a concentration of 2 mg ml−1 for 4 h at 37 °C. Aliquots were collected after 0, 0.5, 1, 2 and 4 h, and 200 μl of acetonitrile was added to each sample (100 μl) and incubated for 10 min at 4 °C. Samples were then processed in an AB SCIEX QTRAP 5500 system. The column used was an Agilent ZORBAX Eclipse XDB-C18 (3.5 μm, 2.1 mm × 150 mm). The mobile phases used were A (100% water with 0.1% v/v formic acid) and B (100% acetonitrile with 0.1% v/v formic acid), Fisher optima grades. Measurements were made by multiple reaction monitoring (MRM). The percentage of remaining undamaged peptide was calculated by integrating the AUC related to the peptide at timepoint zero. The time gradient for mobile phase composition is listed in Supplementary Table 7.
Bacterial resistance development assaysIn wells of a 96-well polypropylene flat-bottom plate, 5 µl of the overnight bacterial culture was added to 100 μl of AMPs/antibiotic solutions in MHB at 105 c.f.u.s per well. Plates were incubated for 20–24 h at 37 °C. The MIC, the lowest concentration of peptide/antibiotic that caused no visible bacterial growth, was determined for each bacterial species. Thereafter, 5 µl of the growth at the 0.5-fold MIC suspension was added to a fresh medium containing AMPs/antibiotics at 105 c.f.u.s per well, and these mixtures were incubated as described above. This was repeated for 20 passages.
SEM measurementStrains were grown to the exponential phase. The bacterial suspensions (108 c.f.u.s ml−1) were co-cultured with AMPs at final concentrations of 100 µg ml−1 at 37 °C for 24 h, and untreated cells were used as control. The specimens were observed using the FE-SEM Regulus8100 (Hitachi) scanning electron microscope.
Detection of peptide-induced membrane permeabilitySingle colonies of strains were inoculated and cultured to the exponential phase, followed by three washes with 10 mM PBS (pH 7.0) and adjustment of OD625 values to 0.08~0.13 with PBS. Subsequently, 150 μl of the bacterial suspension was incubated at 37 °C with 50 μl AMP (dissolved in PBS) for 2 h. PI was added at a final concentration of 50 μg ml−1, and the mixture was incubated in the dark at 37 °C for 30 min. Thereafter, bacterial suspensions were centrifuged (4 °C 1,825 × g, 10 min) and washed with PBS twice. All experiments were performed with three independent replicates. The treated cells were examined by flow cytometry (ThermoFisher Attune NxT). Data were analysed using FlowJo (v.10.8.1).
DiSC3-(5) assayCRAB QLH-2022-637 and MRSA QLH-2022-718 were grown at 37 °C with agitation until they reached mid-log phase. The cells were then centrifuged and washed twice with washing buffer (20 mmol l−1 glucose, 5 mmol l−1 HEPES, pH 7.2) and resuspended to an OD600 of 0.05 in the same buffer containing 0.1 mol l−1 KCl. The cells (100 μl) were then incubated for 15 min with 20 nmol l−1 of DiSC3-(5) until the reduction of fluorescence stabilized, indicating the incorporation of the dye into the bacterial membrane. Membrane depolarization was then monitored by observing the change in the fluorescence emission intensity of the membrane potential-sensitive dye, DiSC3-(5) (lex = 622 nm, lem = 670 nm), after the addition of the peptides (100 μl solution at MIC values). Relative fluorescence was calculated as:
$$}\; }\left(\% \right)=\frac\left(\rm\right)}}\rm}}\times 100 \%$$
(17)
RNA sequencing and analysisE. coli ATCC25922 was treated with AMP (1-fold MIC) for 2 h with six replicates, while PBS was used in the control group. RNA sequencing was conducted using the Illumina NovaSeq platform. HTSeq v.0.6.1 was used to count the read numbers mapped to each gene, and then the fragments per kilobase of transcript per million fragments mapped (FPKM) of each gene was calculated. Differential expression analysis was performed using the DESeq R package. PCoA and PERMANOVA were conducted using adonis2 (vegan R package).
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Comments (0)