
Remember me

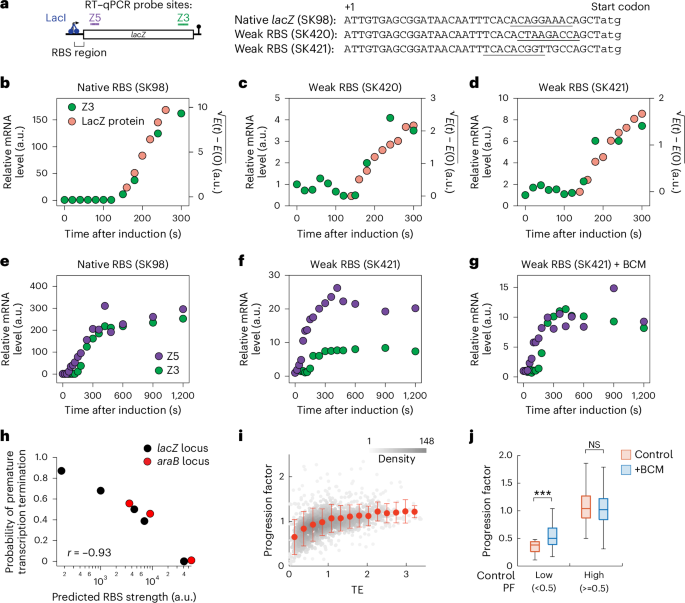
Participant 1 (P1) is a male diagnosed with HIV-1 in 2019, at which time the plasma HIV-1 RNA was 1,240,000 copies per ml and the CD4+ T cell count was 8 cells per µl. He responded to initial ART, but despite increasing CD4+ T cells, maintained persistent low-level viraemia (100–1,000 copies per ml) for the 4.5-year study period despite a regimen switch, therapeutic drug monitoring with dose optimization, and the absence of relevant resistance mutations (Fig. 1a).
Fig. 1: Failure to suppress viraemia is characterized by diverse virus in plasma and high infected-cell frequency.
a,b, Plasma HIV-1 RNA and CD4+ T cell counts over time for P1 (a) and P2 (b). Numbers above squares represent CD4+ T cell percentages. The dotted line at 20 copies per ml represents the current limit of detection for the clinical HIV-1 viral load assay. Data are for periods with optimal adherence. For P2, there was a preceding period of suboptimal adherence (see text and Extended Data Fig. 1a). c,d, Neighbour-joining phylogenetic trees of p6-RT single genome sequences obtained from plasma viral RNA for P1 (c) and P2 (d). Phylogenetic tree tip labels are colour coded according to the plasma collection timepoint in a. Phylogenetic trees are rooted to HXB2, and HIV-1 coordinates refer to the HXB2 reference genome. Tree nodes with bootstrap values >80 are marked with asterisks. e, Intact proviral DNA frequencies as measured by the IPDA. The percentage of proviruses classified as intact by the IPDA is shown on the top. Asterisks represent analyses performed on total white blood cells that were corrected on the basis of the CD4+ T cell percentage at the time of sampling. f, Infectious units per million (IUPM) CD4+ T cells as measured by the quantitative viral outgrowth assay. g, IUPM to IPDA intact ratio as measured by dividing the IUPM by the closest IPDA timepoint value. TAF, tenofovir alafenamide; FTC, emtricitabine; BIC, bictegravir; DRV/c, darunavir/cobicistat; b.i.d., bis in die (twice daily); DRV/r, darunavir/ritonavir; DTG, dolutegravir.
Participant 2 (P2), a female, was diagnosed with HIV-1 in 2003, with a viral load of 23,343 copies per ml and a CD4+ T cell count of 301 cells per μl. P2 initially achieved viral suppression with ART but experienced intermittent adherence over the next several years, resulting in virological failure and CD4+ T cell depletion (Extended Data Fig. 1a; CD4+ T cell nadir of 4 cells per μl). Following hospitalization due to opportunistic infection, a new ART regimen was initiated. Despite adherence for more than 9 years, virologic suppression was not achieved (Fig. 1b). Both participants recovered CD4+ T cell counts (peak CD4+ T cell count of 555 and 1,074 cells per µl, respectively; Extended Data Table 1) despite the low nadirs (Fig. 1a,b). Neither participant has HLA alleles associated with elite control or rapid progression. Since most PWH achieve viral suppression (<50 copies HIV-1 RNA per ml plasma) within 12 weeks of ART initiation34,35,36,37, the persistence of detectable viraemia after years of treatment with optimal adherence represents a rare and troubling clinical outcome.
Virus populations in plasma are diverse and show no evidence of evolutionTo investigate the cause of the NSV, we analysed longitudinal plasma HIV-1 RNA sequences (pro-pol). In both participants, the plasma virus was highly heterogeneous, in contrast to previous reports of residual viraemia from PWH on ART, which shows that viraemia is typically dominated by few predominant plasma clones (Fig. 1c,d and Extended Data Fig. 1b)18,21,25,26,27.
Plasma virus sequences from both participants lacked the temporal structure characteristic of ongoing viral evolution and showed no accumulation of diversity, divergence, or shift in viral populations over time (Extended Data Fig. 1c and Methods)38. Notably, we found no drug-resistant mutations affecting the concurrent regimen, a result confirmed through clinical genotyping. These results demonstrate that NSV was not driven by ongoing replication or selection for drug resistance.
Large pool of infected cells contributes to persistent viraemiaGiven the heterogeneity of plasma virus, we hypothesized that viraemia in the absence of ongoing replication might reflect an unusually high frequency of infected CD4+ T cells39. The intact proviral DNA assay (IPDA)40 showed that the frequency of proviruses classified as intact in P1 and P2 was markedly higher than previously reported in 81 PWH on ART (151 copies per 106 CD4+ T cells; Fig. 1e)41. Despite the high frequency of intact proviruses by IPDA, the frequencies of cells with inducible, replication-competent proviruses detected by the quantitative viral outgrowth assay (QVOA) were comparable to those of previous studies: 3.18 infectious units per million (IUPM) CD4+ T cells for P1 and 1.44 IUPM for P2 (Fig. 1f)42. The ratio of IUPM to intact proviruses—a metric used to estimate reservoir inducibility43—was much lower than previously reported in PWH on ART (Fig. 1g)40, suggesting that a much smaller than normal fraction of the proviruses detected in the IPDA could produce infectious virus.
Most proviruses in P1 and P2 have shared deletions affecting key genesTo explain the discrepancy between IPDA and QVOA measurements, we sequenced near full-length proviral genomes from P1 and P2. In each participant, >90% of the proviral sequences shared identical large deletions but, surprisingly, were divergent elsewhere in the genome (Fig. 2).
Fig. 2: Near full-length sequencing reveals dominant proviral populations with unique deletion signatures and diverse mutations across the viral genome.
a,e, Left: neighbour-joining phylogenetic trees of near full-length proviral sequences obtained by single genome sequencing from primary CD4+ T cells, rooted to HXB2. The colour of each branch tip indicates sampling time as in Fig. 1a,b. Dashed branches indicate sequences with hypermutation. Right: highlighter plot with black lines representing nucleotide changes compared to the top sequence. Grey vertical bars represent deletions compared to HXB2. Highlighted areas represent recurrent deletion patterns of 1,417 nt, 313 nt (P1) and 270 nt (P2). IPDA primer probe regions are highlighted at the top. Green arrowheads point to sequences with significant G→A hypermutation. b,f, Mapped sequences of prominent deletion signatures found in majority of proviruses in P1 and P2 compared to HXB2. c,g, Dot plots representing the number of mutations between each near full-length sequence and the majority consensus sequences. Data represent mean ± s.d. (c) n = 32 and (g) n = 65. d,h, Histogram (bin width of 5) representing the nucleotide differences in unique proviruses from each participant. Dashed line represents the mean number of nucleotide differences between all unique proviral sequences.
In P1, 50/54 sequences contained the same set of two deletions of 1,417 and 313 nucleotides (nt) in exactly the same positions. The 1,417-nt and 313-nt deletions affected the ORFs of vif, vpr, tat, rev, vpu and env (Fig. 2a,b). These deletions would likely preclude further replication, yet the proviruses harbouring these exact two deletions were remarkably diverse elsewhere in the genome (Fig. 2a). To assess the diversity within the proviral sequences, we first compared the number of nucleotide changes between each provirus and a majority consensus sequence containing the deletions to estimate viral diversification. The mean distance to the majority consensus sequence was 81 nt (Fig. 2c). Next, we measured the average pairwise distance (APD) to find the genetic diversity within the proviral population. The APD between unique near full-length proviral sequences was 1.38%, suggesting that, on average, there were 125 nucleotide differences between unique sequences (Fig. 2d). Furthermore, some of the proviruses with the recurring deletions also had signatures of APOBEC3G/F-mediated hypermutation (Fig. 2a), indicating that viral RNA harbouring these deletions entered a target cell where it underwent hypermutation during reverse transcription.
In P2, we found a 270-nt deletion impacting the reading frames of vpr, tat, rev and vpu in 64/65 sequences (Fig. 2e,f). Given the critical roles of Tat and Rev in the virus life cycle, this deletion should preclude replication of the deleted virus. As with P1, proviruses with this deletion showed significant diversity elsewhere in the viral genome. The mean distance from the majority consensus sequence was 109 nt (Fig. 2g). The APD between near full-length proviral sequences with this deletion was 1.81%, corresponding to an average of 164 nucleotide differences (Fig. 2h). The deletions in proviruses from P1 and P2 did not impact the binding of the IPDA primers and probes (Fig. 2a,e), explaining the high frequency of proviruses classified as intact (Fig. 1e).
Identical sequences harbouring fatal deletions typically indicate proliferation of CD4+ T cell clones carrying defective proviruses16,18,25,44,45,46, since these deletions render the virus unable to replicate8,9,21. However, the high diversity of proviruses with the same fatal deletions is inconsistent with clonal expansion and cannot be explained by errors made by human RNA polymerase II, or by PCR and sequencing errors. The finding of nucleotide differences in viral genomes carrying the same fatal deletions implies that defective viral RNA was packaged into virions, entered a target cell, underwent reverse transcription leading to additional mutations, and was then integrated into the host genome.
To quantify proviruses harbouring these deletions, we designed digital PCR (dPCR) assays with primers and probes detecting each deletion separately (Fig. 3a,b). Assay specificity was validated using genomic DNA from other PWH; only DNA from P1 and P2 yielded positive signals from deletion-specific dPCR assays (Extended Data Fig. 2a). In both participants, we found that >80% of proviruses contained these deletions, and the proportion of proviruses containing the deletions remained stable over time on ART (Fig. 3c). Taken together, these results show that most infected cells contained these deletions.
Fig. 3: Defective proviruses dominate the proviral landscape and contribute to NSV.
a, Location of primers (arrows) and probes (rectangles with vertical bars) to specifically quantify deletion signatures in viral RNA and DNA. Probes span the deletion region. b, Representative two-dimension plots of dPCR showing duplex amplification of intact proviruses and proviruses of interest by deletion-specific assays. c, Longitudinal quantification of proviruses with specific deletions of interest. Data are mean of technical triplicates (n = 3). Asterisks represent analyses performed on total white blood cells that were corrected on the basis of the CD4+ T cell percentage at the time of sampling. d, CD4+ T cells from P1 and P2 were cultured for 48 or 72 h in the presence of emtricitabine (FTC), tenofovir disoproxil fumarate (TDF), dolutegravir (DTG) and anti-CD3/CD28 beads. The virion-associated RNA in the supernatant was measured by RT–dPCR. Error bars represent s.e.m. Pie chart shows percentage of HIV-1 RNA copies with deletion normalized to the total number of copies. e, Plasma virion-associated RNA was measured by RT–dPCR. Data are mean ± s.e.m. performed in at least technical triplicates (n = 3–8). Pie chart shows percentage of HIV-1 RNA copies with deletion normalized to the total number of copies. For P1, total proviruses were quantified by measuring the highly conserved region LTR-gag. For P2, proviruses without the 270-nt deletion were measured using a primer–probe set inside of the deletion (WT-270).
Proviruses with recurrent deletions can express viral RNA and contribute to persistent viraemiaGiven that some defective proviruses can express HIV-1 RNA and produce viral proteins in vivo21,23,24, we evaluated whether proviruses harbouring these fatal deletions could produce virions that package the defective viral RNA. We stimulated CD4+ T cells from P1 and P2 with anti-CD3/CD28 beads in the presence of antiretroviral drugs and quantified supernatant viral RNA using the RT–dPCR assay described above. In P1, we found that, on average, 55% of virion-associated RNA contained either the 1,417-nt or 313-nt deletion (Fig. 3d). In P2, 13% of the virion-associated RNA in the supernatant contained the 270-nt deletion (Fig. 3d).
Given that infected cells from both participants could be induced to make virions packaging these defective RNAs, we determined whether cells with these proviruses contributed to the NSV. Using the same digital PCR assays, we found that in both participants, ~50% of virion-associated HIV-1 RNA in plasma harboured these deletions (Fig. 3e). Given that viral loads measured with clinical assays were in the range of 102–103 copies per ml, it is possible that viraemia would remain detectable even with defect-specific assays.
To determine when defective proviruses arose, we analysed longitudinal pre-treatment samples for P2. The Δ270 deletion was absent in proviruses and in plasma at 2 years and at 6 years post diagnosis, respectively, but was present after ART initiation (Extended Data Fig. 2b–d). These results suggest that the Δ270 deletion arose between 6 and 12 years post diagnosis, during the prolonged period of suboptimal ART adherence.
We also sequenced QVOA supernatants to determine whether proviruses with these specific deletions could produce infectious virus. Despite an abundance of cells carrying defective proviruses, most viruses detected had intact genomes (Extended Data Fig. 3a,b). However, virions packaging defective RNA were also detected, consistent with our results from ex vivo stimulation of infected CD4+ cells.
Together, these results show that although the recurring deletions are incompatible with replication, viral RNA from the dominant deleted proviruses can be packaged into virions and contribute to persistent viraemia.
Molecular clones with these deletions show significantly reduced virion productionTo investigate the impact of deletions on replicative fitness and virus production, we introduced the recurring deletions into a reference proviral construct (NL4-3) and transfected HEK293T cells (Fig. 4a)47. As expected, deletions in key HIV-1 genes resulted in an almost complete loss of virus production (>100,000-fold p24 reduction) compared to the wild type (Fig. 4b). We also stained transfected HEK293T cells with multiple broadly neutralizing antibodies (bNAbs) to assess Envelope (Env) expression (Fig. 4c and Extended Data Fig. 4). Consistent with p24 enzyme-linked immunosorbent assay (ELISA) results, the percentage of HEK293T cells expressing Env was not statistically different from that of cells transfected with a construct lacking env (NL4-3-ΔEnv; NSP > 0.01; Fig. 4d).
Fig. 4: Deletions found in proviruses abolish virus production in vitro.
a, Prominent deletions found in proviruses were introduced into an NL4-3 expression plasmid by site-directed mutagenesis. Arrows represent orientation of the provirus within the expression plasmid. b, Virus produced upon HEK293T transfection was pelleted by ultracentrifugation, and p24 was measured by ELISA. Lower limit of detection (LoD) was 0.625 ng ml−1. Data represent mean ± s.d. c, Representative flow cytometry plots of the surface staining of transfected HEK293T cells with viability dye and bNAbs (VRC01 and 3BNC117). d, Surface staining of HIV-1 Env with bNAbs (VRC01 and 3BNC117) on HEK293T cells 24 h after transfection. Data represent mean ± s.e.m. (b,d) Statistical significance between conditions was determined using one-way analysis of variance (ANOVA). ****P < 0.0001, NSP > 0.01.
These observations show that virion-associated RNA in the supernatant of activated infected cells and in plasma contained the same deletions found in the proviral sequencing. However, the deletions resulted in abrogated virion production when introduced into replication-competent molecular clones. To address this apparent discrepancy, we investigated alternative mechanisms for the dissemination of these proviruses.
Dissemination of defective proviruses via superinfectionWe hypothesized that intact HIV-1 virions could superinfect cells carrying proviruses with the recurrent deletions and drive the production of infectious viral particles packaging defective genomic RNA. In superinfected cells, viral proteins can be produced from the intact provirus, but genomic RNA from both defective and intact proviruses compete for packaging into the virions48. This allows production of infectious particles that have packaged the defective viral RNA. Upon infection of new cells, the defective RNA genome undergoes reverse transcription, accumulates additional mutations and then integrates into the host cell genome. In this manner, defective proviruses carrying the same fatal deletions could continue to accumulate a diverse set of additional mutations. The competition between replication-competent and defective viral genomes for packaging in co-infected cells could result in viral interference, consistent with DIPs30,31. Therefore, we hypothesized that viruses with the recurrent deletions could interfere with WT (intact) virus replication. To assess this hypothesis, we generated cell lines containing NL4-3-based reporter constructs (NL4-3-ΔNef-BFP) modified to include the deletions found in each participant (P1: Δ1417Δ313, P2: Δ270; Methods and Extended Data Fig. 5a) and superinfected them with an infectious WT reporter virus (NL4-3-ΔNef-RFP) (Fig. 5a,b and Extended Data Fig. 5b). The spread of WT virus in cells carrying the defective proviruses was significantly reduced compared to WT virus spreading in mock-transduced SupT1 cells, suggesting that these defective proviruses interfere with WT virus replication (Fig. 5b). We quantified the infectivity of WT virus by using the supernatant from day 3 of culture to infect new SupT1 cells in a single-round infectivity assay (Fig. 5c)49. Supernatant from cells harbouring defective proviruses contained significantly less infectious WT virus than supernatant from control cells, further confirming viral interference (Fig. 5c). In addition, this single-round infectivity assay showed that the defective genomes were efficiently packaged and transmitted. When supernatant from the superinfected cultures was used to infect new SupT1 cells, >40% of target cells were infected with the defective virus (BFP+) (Δ1417Δ313: 43%, Δ270: 62%) (Fig. 5d).
Fig. 5: Cell culture model to assess superinfection in vitro.
a, Schematic for assessment of superinfection in vitro. Top: SupT1 cells are infected with NL4-3-ΔNef-RFP WT virus. Bottom: model cell lines transduced with reporter defective viruses representing P1 (Δ1417Δ313) or P2 (Δ270) are infected with NL4-3-ΔNef-RFP WT virus. Virions produced by these superinfected cells may package RNA from the defective (blue) or WT (red) viral genome. b, Frequency of cells infected with the WT virus (% RFP+) measured over time. Data represent mean ± s.d. c, Viral supernatant from day 3 of infection of each cell line was used to infect target SupT1 cells to determine the viral titre of WT virus after a single round of infection. WT viral titre was normalized to virus from the WT SupT1 cell line. Data represent mean ± s.e.m. d, Viral supernatant from day 3 of infection of each cell line was used to infect SupT1 cells to determine the fraction of transmitted provirus after a single round of infection as measured by flow cytometry. e, Schema for assessment of conditional replication in vitro. Model cell harbouring reporter defective viruses are either mock infected or infected with WT virus. After 3 days of culture, the resulting supernatant is used to infect target SupT1 cells. f,g, Frequency of target SupT1 cells infected with defective provirus measured over time. Data represent mean ± s.d. h, Top: schema for three-colour experiment to estimate R0 in vitro. WT SupT1 or SupT1-Δ1417Δ313-BFP are infected with WT virus. After 2 days, the cells were co-cultured with excess GFP+ SupT1 target cells. Bottom: frequency of target GFP+ SupT1 target cells infected with either WT virus (red) or defective virus (blue). R0 values ± s.e.m. for both the WT virus (R0WT) and defective virus (R0DEF) are listed at the bottom. Data represent mean ± s.d. (b–d) n = 4 for all conditions assessed. (f–h) n = 6 for all conditions assessed. (b,c) Statistical significance between cell lines was determined using one-way ANOVA. ****P < 0.0001, NSP > 0.05.
To validate that the defective proviruses conditionally replicate in the presence of WT virus, we collected supernatants from either the deleted-variant cell lines alone or from the deleted-variant cell lines that had been infected with WT virus for 3 days. These supernatants were used to infect SupT1 cells to assess transmission of the defective provirus (BFP+) (Fig. 5e). Supernatant from the cells harbouring defective proviruses alone was largely non-infectious. However, when first superinfected with WT virus, the subsequent transmission of defective genomes to target cells increased dramatically (P1: >170-fold, P2: >3.69-fold; Fig. 5f,g). Thus, defective proviruses transmit their genome to target cells when superinfected with WT virus. These results demonstrate that the recurrent proviral deletions in P1 and P2 interfere with WT virus replication and conditionally replicate in the presence of WT virus, hallmarks of DIPs50.
Given that the deletions found in P1 (Δ1417Δ313) resembled the engineered therapeutic interfering particle (TIP) described in ref. 30 (for example, deletion of tat, rev, vpu and env, and retention of cPPT) (Extended Data Fig. 5c), and considering the ability of defective genomes to integrate into target cells (Fig. 5d), we hypothesized that the defective proviruses found in P1 functioned as a TIP30. For a DIP to be considered a TIP, it must be mobilized with a basic reproductive ratio, R0DEF, of >1, meaning that the defective provirus can give rise to virions that infect more than one target cell. To measure the R0 of the defective variant, we implemented a three-colour assay (Fig. 5h, Methods and Extended Data Fig. 5d)30. The R0DEF represents the average number of target cells infected with the defective virus resulting from WT virus superinfection of cells carrying the deleted provirus. Notably, the R0DEF in the Δ1417Δ313 reporter cell line was 1.86 (Fig. 5h). This result is consistent with the spread of the defective viruses in vivo. As expected, the assay showed interference of the WT virus, significantly reducing the R0WT from 6.40 in the WT SupT1 cells to 0.84 in the Δ1417Δ313 reporter cells (Fig. 5h; P = 0.0002). The low R0DEF suggests that although viral interference can be detected in vitro (Fig. 5b), the interference does not affect WT viral replication and pathogenesis in vivo to a clinically significant extent, as evidenced by P1’s high initial viral load and extremely low CD4 nadir at the time of ART initiation.
Superinfected CD4+ T cells carrying both intact and defective proviruses can be detected ex vivoTo show that defective proviruses found in P1 and P2 were mobilized—before the introduction of effective ART—by superinfection with intact proviruses, we looked for cells carrying multiple proviruses in primary CD4+ T cells from P1 and P2. We modified the HIV-flow assay51 to sort single p24+ cells into individual wells. We then lysed the cells and distributed the cell lysate over 6 or 12 wells, resulting in a >80% probability that, if two proviruses were present, they would be separated into different wells (Fig. 6a and Extended Data Fig. 6a). Lastly, we amplified and sequenced the near full-length proviral genomes52. Multiple approaches were used to confirm sorting accuracy in this experiment (Methods and Extended Data Fig. 6b–d). Furthermore, in a recent study, Dufour and colleagues did not find any evidence of cells infected with multiple proviruses from 305 single-sorted p24+ cells from 6 PWH on suppressive ART52.
Fig. 6: Single p24+ cell sequencing identifies dually infected cells in vivo.
a, Experimental design to characterize p24+ cells and detect multiple integrated proviruses. Sorted p24+ cells are lysed and spread over multiple wells. Near full-length proviral amplification, agarose gel electrophoresis and sequencing are conducted on the cell lysate. Example agarose gel electrophoresis is shown. b,c, Neighbour-joining phylogenetic tree of near full-length proviral genomes from p24+ cells for P1 (b) and P2 (c). Highlighter plot with black lines represent nucleotide changes compared to the top sequence. Grey boxes represent deletions compared to the top sequence of each tree. Tree nodes with bootstrap values >80 are marked by asterisks. 5′ internal deletions are boxed in black and annotated. Common deletions found in P1 and P2 are boxed and annotated. Phylogenetic tree tips with grey boxes represent single provirus found from a sorted cell. Coloured boxes represent multiple proviruses found from a single sorted cell, probably representing multiple integrated proviruses within one cell. Coloured arrowheads point to multiple proviruses found in a single sorted cell.
For P1, we analysed ~13 million CD4+ T cells and recovered 7 sequences from the p24+ sorted cells (0.54 proviruses per 106 CD4+ T cells) (Fig. 6b). Notably, we identified one cell that had two integrated proviruses: one with the 1,417-nt and 313-nt deletions and one nearly intact provirus with a small 5′ deletion, demonstrating the presence of superinfected cells in vivo (Fig. 6b, colour boxed). For P2, we analysed ~13 million CD4+ T cells and recovered 4 sequences from the sorted cells (0.30 proviruses per 106 CD4+ T cells) (Fig. 6c). From two cells, we identified one intact provirus in each. Notably, in one p24+ sorted cell, we detected two proviruses: one carrying the 270-nt deletion and one that was intact across the entire viral genome (Fig. 6c, coloured boxes).
Together, these results provide direct evidence of persistent infected cells carrying two proviruses and support our in vitro studies showing that superinfection allowed the mobilization of defective proviruses and the accumulation of mutations.
Comments (0)