
Remember me
Hepatocellular carcinoma (HCC), the most common primary liver malignancy, poses a major challenge in clinical medicine.[1] It is closely linked to chronic liver disease and is the second leading cause of cancer-related death in China, with both incidence and mortality steadily increasing.[2] The lack of highly sensitive and specific clinical biomarkers limits the effectiveness of current early-stage screening strategies. Despite advances in treatment, including surgery, chemotherapy, and radiotherapy, HCC prognosis remains poor, with overall survival rates ranging from 25% to 39%.[3] Therefore, the identification of novel targeted therapies and tumor markers for the early HCC detection, along with the development of more effective treatment regimens, is urgently needed to improve patient outcomes.
Ferroptosis, a recently identified form of regulated cell death, is characterized by the iron-dependent accumulation of lipid peroxides, leading to oxidative membrane damage.[4] Ferroptosis, unlike apoptosis, necrosis, or autophagy, is driven by distinct genetic and biochemical pathways, primarily involving dysregulation of glutathione metabolism and glutathione peroxidase 4 (GPX4) activity.[5] Growing evidence indicates that ferroptosis plays a crucial role in tumor suppression and may offer new therapeutic opportunities for cancer treatment. In HCC, ferroptosis has been shown to inhibit tumor progression and enhance chemosensitivity, highlighting its potential as a therapeutic target.[6] The link between ferroptosis and HCC pathogenesis underscores the importance of investigating novel regulators of this cell death pathway.[7]
Progestin and adipoQ receptor 3 (PAQR3), also known as Raf kinase trapping to Golgi, is a member of the PAQR protein family, which includes a class of recently revealed receptors and a transmembrane protein located in the Golgi apparatus.[8] PAQR3 is a newly identified gene with multiple potential tumor-suppressive functions, including inhibition of cell migration, proliferation, sprouting, and endothelial cell angiogenesis through downregulation of the MAPK signaling pathway.[9] Moreover, it may function as a candidate inhibitor of gastric adenocarcinoma through modulation of the transforming growth factor-b (TGF-b)/ Smad pathway.[8] Emerging evidence indicates that PAQR3 is implicated in numerous human cancers, such as HCC and clear-cell renal cell carcinoma.[10] Notably, prior research has suggested that PAQR3 has a vital role in HCC progression.[11] However, its potential contribution to ferroptosis regulation in HCC remains unexplored, and the full implications of altered PAQR3 expression in HCC remain unclear.
Multiple cellular genes and signaling pathways have been associated with HCC pathogenesis, including the TGF-b signaling pathway.[12] TGF-b, a ~25 kDa polypeptide cytokine belonging to the TGF family, exhibits diverse biological activities and regulates various cellular processes through autocrine or paracrine mechanisms, such as angiogenesis, apoptosis, proliferation, adhesion, and cellular differentiation.[13] Moreover, aberrant activation of the TGF-b pathway is among the most frequently altered signaling pathways in several tumors and plays a critical role in tumorigenesis and progression.[14] Nevertheless, the impact and mechanisms of altered PAQR3 expression and TGF-b pathway activity in HCC remain incompletely understood. Therefore, this study aimed to examine PAQR3 expression in primary HCC and to investigate its biological effects and underlying mechanisms in HCC metastasis, with a particular focus on its potential role in ferroptosis regulation.
MATERIAL AND METHODS Patients and tissue samplesHCC specimens and their corresponding adjacent non-cancerous tissues, located at least 2 cm from the tumor site, were obtained from HCC patients who previously underwent hepatectomy at the General Hospital of Tianjin Medical University. Histopathological examination confirmed the diagnosis, and none of the patients underwent radiotherapy or chemotherapy before surgery. A total of 106 paired formalin-fixed paraffin-embedded specimens were subjected to PAQR3 immunohistochemistry (IHC) and immunoreactivity score (IRS) scoring. For each specimen, three non-overlapping high-power fields (×200) were evaluated, and the scores were averaged. Two board-certified pathologists, blinded to clinical data, independently scored the slides, and any discrepancies were resolved by consensus.
All patients provided written informed consent before sample collection. The study protocol was reviewed and approved by the Institutional Ethics Committee of Tianjin Medical University (Approval No.8217102738). All procedures involving human participants were performed in accordance with the ethical standards of the Institutional Research Committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
IHCBefore the assay, liver tissues were sectioned at 4 μm thickness. The samples were rehydrated through graded ethanol concentrations from 50% to 100% after deparaffinization with xylene. Antigen retrieval was performed using ethylenediaminetetraacetic acid. Sections were then blocked with 10% normal goat serum (Solarbio, SL038) for 1 h at room temperature to reduce non-specific binding. Subsequently, the sections were incubated overnight at 4°C with the PAQR3 primary antibody (1:200; Affinity Biosciences, DF4988, China), followed by incubation with an HRP-conjugated secondary antibody (1:500; Servicebio, GB23303) for 1 h at room temperature. Staining was developed using diaminobenzidine (DAB; Wuhan Golden Wing Industry and Trade Co., Ltd., 91-95-2 Wuhan, Hubei, China). Finally, positive staining was visualized and photographed with an inverted microscope (Leica Microsystems, Wetzlar, Germany). The stained sections were reviewed and scored independently by two pathologists blinded to clinical outcomes. PAQR3 expression was evaluated based on staining intensity (0, negative; 1, weak; 2, moderate; 3, strong) and the proportion of positive tumor cells (0, <5%; 1, 5–25%; 2, 26–50%; 3, 51–75%; 4, >75%). The final IRS was obtained by multiplying the intensity and proportion scores, yielding a range from 0 to 12. For statistical analysis, IRS scores of 0–4 were classified as low expression, and 5–12 as high expression.[15]
Cell culture, treatment, and transfectionHCC cell line Huh-7 (SCSP-526) was sourced from authenticated cell cultures’ national collection, Human Hepatic Cell line 5 (HHL-5) (BFN6072012687) normal human hepatocytes and HCC cell line SNU-449 (CVCL_0454) were sourced from American Type Culture Collection, and Hep3B (CL-0102) was purchased from Procell (Wuhan, China). All cells were maintained in RPMI-1640 medium (Thermo Fisher Scientific, Inc., Waltham, Massachusetts, USA) in 5% CO2 at 37°C, added with 10% fetal bovine serum (FBS) and 1% streptomycin/penicillin.
Hep3B cells were treated with 2.5 μM of the TGF-b1 inhibitor SB-431542 (AbMole Bioscience, Inc., M57235, Houston, Texas, USA) for 24 h, or with 10 ng/mL TGF-b1 (AbMole Bioscience, Inc., M9391, Houston, Texas, USA) for 24 h.
A PAQR3 overexpressing plasmid DNA (Oe-PAQR3) was used to induce PAQR3 overexpression in Hep3B cells. Cells were transfected with 100 nM of the recombinant constructs using Lipofectamine® 2000 (Thermo Fisher Scientific, Inc., 11668019, Waltham, Massachusetts, USA) for 48 h at 37°C. After a 48-h incubation at 37°C, the transfection efficiency of Oe-PAQR3 was evaluated, and the transfected cells were subsequently used for further experiments.
All HCC cell lines (Huh-7, Hep3B, SNU-449) and the normal hepatocyte line HHL-5 used in this study were authenticated by short-tandem repeat (STR) profiling (performed by [specify service provider, e.g., China Center for Type Culture Collection, Wuhan, China]) to verify their identity and to exclude cross-contamination. Mycoplasma contamination was routinely monitored using a polymerase chain reaction-based mycoplasma detection kit (e.g., Venor®GeM, Minerva Biolabs, Berlin, Germany), and all cell lines tested negative before experimental use.
Western blotting (WB)Proteins were isolated carefully from sample cells using RIPA lysis buffer (Shenzhen Ziker Biotechnology Co., Ltd., ZK-L2341, Shenzhen, Guangdong, China) and quantified with a bicinchoninic acid protein assay kit (Shanghai Kuaibo Biotechnology Co., ZK-L2356, Ltd., Shanghai, China) according to the standard protocol. After separation by 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, the proteins were then transferred to polyvinylidene fluoride membranes. The membranes, blocked with 5% non-fat milk, were incubated overnight at 4°C with primary antibodies (Abcam plc, Cambridge, UK) at the following dilutions: GPX4 (1:400, ab262509), SLC7A11 (1:400, ab307601), ACSL4 (1:1500, ab269613), PAQR3 (1:100, ab236798), E-cadherin (1:1000, ab314063), Vimentin (1:2000, ab92547), N-cadherin (1:1000, ab76011), TGF-b1 (1:1000, ab215715), p-Smad3 (1:2000, ab74062), p-Smad2 (1:1000, ab184557), Smad2 (1:2000, ab40855), Smad3 (1:500, ab40854), and b-actin (1:2500, ab8226). Subsequently, the membranes were incubated with horseradish peroxidase-conjugated secondary anti-rabbit antibody (Proteintech Group, Inc., S0001, Rosemont, Illinois, USA) at room temperature. Protein bands were detected utilizing an Image Analysis System (Bio-Rad Laboratories, Inc., ChemiDoc™, Hercules, California, USA) with an Enhanced Chemiluminescence Fluorescence Detection kit (Cytiva, Uppsala, Sweden).
Wound healing assayCells were seeded into 24-well plates, which were then cultured overnight. When confluency reached 95%, a linear scratch was made using a 200 μL pipette tip. Images of the wound were captured at 0 and 24 h using a microscope (Leica, Mateo FL, Germany), and the wound closure rate was quantitatively assessed using ImageJ software.
Transwell assayA 200 μL cell suspension in serum-free DMEM was added into the upper chamber of the Transwell, while 600 μL of DMEM supplemented with 2% FBS was placed into the lower chamber. Following a 24 h incubation at 37°C, the cells were fixed using 4% paraformaldehyde. Cells adhering to the underside of the membrane were then stained with crystal violet solution for 15 min (Beyotime Institute of Biotechnology, Y268091, Haimen, Jiangsu, China), and images were randomly captured using an inverted microscope (Leica Microsystem, Mateo FL, Wetzlar, Germany).
Thiobarbituric acid reactive substance (TBARS) assayLipid peroxidation in HCC cells following the indicated treatments was assessed using a TBARS Assay Kit (Elabscience Biotechnology Co., Ltd., E-BC-K298-M, Wuhan, Hubei, China). Briefly, cells were treated with butylated hydroxyanisole (BHA; 10 μL; 500 mM) and trichloroacetic acid (0.5 mL; 15%), followed by centrifugation at 10,000 × g for 10 min at 4°C. The obtained supernatant was thoroughly mixed with 0.5 mL of 0.375% and boiled for 10 min. After cooling, TBARS levels were determined at 532 nm using a microplate reader (Bio-Tek Instruments, Inc., Varioskan ALF, Winooski, Vermont, USA).
Lipid reactive oxygen species (ROS) stainingROS levels in HCC cells were assessed using C11-BODIPY 581/591 (Thermo Fisher Scientific, Waltham, D3861, Massachusetts, USA). The cell culture medium was supplemented with C11-BODIPY 581/591 to a final concentration of 5 μM. Following incubation in the dark at 37 °C for 30 min and subsequent PBS washes, the cells were imaged using a fluorescence microscope (Leica Microsystems, Mateo FL, Wetzlar, Germany).
Intracellular iron measurementFollowing the indicated treatments, HCC cells were incubated with 10 μM Phen Green SK (PGSK, Glpbio, GC40243, Houston, Texas, USA) for 10 min. Excess PGSK was detached and carefully eliminated by washing the cells twice with PBS. The cells were then trypsinized, resuspended in PBS containing 5% FBS, and ferrous iron levels were determined using a flow cytometer (Beckman Coulter, Inc., CytoFLEX S, Brea, California, USA).
Statistical analysisStatistical analyses were performed using GraphPad Prism 8.0 (GraphPad Software, LLC, San Diego, CA, USA). Data are presented as mean±standard deviation. Comparisons between two groups were performed using an unpaired Student‘s t-test, while comparisons among three or more groups were analyzed by one-way analysis of variance, followed by Tukey’s post hoc test for multiple comparisons. Statistical significance was defined as P<0.05. Exact P-values are reported in the Results and figure legends whenever possible; only when P-values were extremely small (< 0.001), thresholds were used for clarity. For paired ordinal data (e.g., IRS for tumor vs. adjacent tissues), the Wilcoxon signed-rank test was applied, and data are displayed as individual values with median and interquartile range unless otherwise stated.
RESULTS PAQR3 expression is significantly reduced in HCC tissuesIHC analysis of 106 paired HCC and adjacent non-tumor tissues revealed markedly lower PAQR3 expression in HCC tissues compared with adjacent counterparts. Representative IHC staining images are shown [Figure 1a]. Quantitative evaluation using the IRS confirmed this observation, demonstrating a significant reduction of PAQR3 expression in tumor tissues relative to paired adjacent tissues (n = 106, ✶✶✶P < 0.001, Wilcoxon signed-rank test; [Figure 1b]).

Export to PPT
Overexpression of PAQR3 inhibits HCC cell metastasisPAQR3 expression in HCC cell lines was measured using quantitative polymerase chain reaction and WB. The results indicated that PAQR3 levels were markedly lower in HCC cell lines compared to HHL-5 normal human hepatocytes (P < 0.001, [Figure 2a and b]). To further explore the mechanism of PAQR3, it was overexpressed through transfection with Oe-PAQR3 [Figure 2c and d]. The effects of PAQR3 overexpression on HCC cell invasion and migration were then examined. As shown in Figure 2e-h, PAQR3 overexpression suppressed both invasion and migration of HCC cells. Furthermore, WB results demonstrated that PAQR3 overexpression led to a marked increase in the expression of the epithelial-mesenchymal transition (EMT)-related protein E-cadherin, while the levels of N-cadherin and Vimentin were notably decreased (P < 0.001, [Figure 2i-l]).
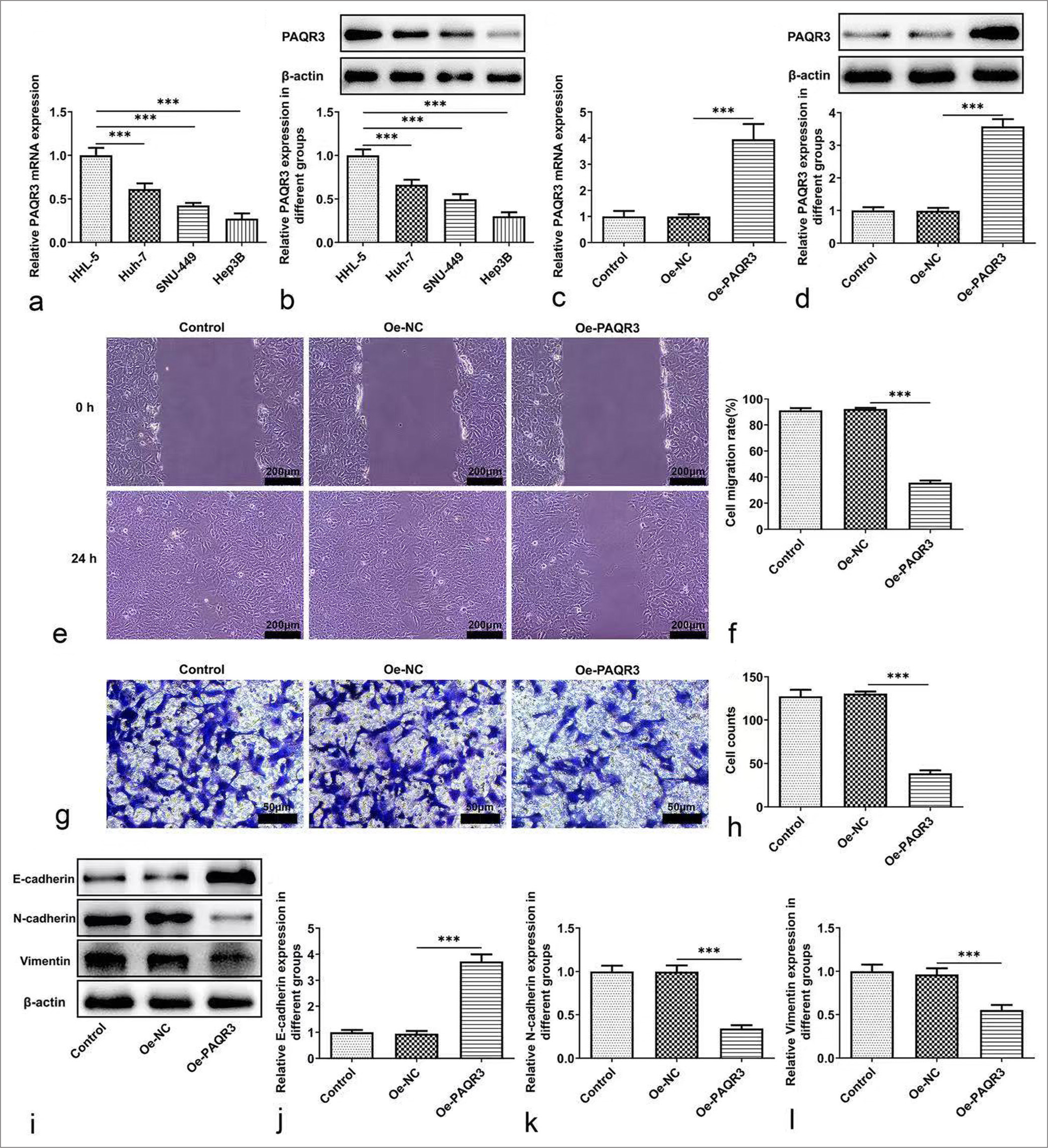
Export to PPT
Overexpression of PAQR3 induces ferroptosis in HCC cellsTo investigate the effect of PAQR3 on ferroptosis in HCC cells, lipid peroxidation was first assessed using the TBARS assay. The results showed that PAQR3 overexpression significantly elevated TBARS production compared with the Control and Oe-NC groups (P < 0.001, [Figure 3a]). Consistently, lipid ROS levels assessed by C11-BODIPY staining revealed a marked increase in oxidized C11 signal and a decrease in non-oxidized C11 signal upon PAQR3 overexpression (P < 0.001, [Figure 3b and c]). Intracellular Fe2+ levels were also significantly elevated in the Oe-PAQR3 group compared with controls (P < 0.001, [Figure 3d and e]). WB analysis demonstrated substantial alterations in ferroptosis-related proteins following PAQR3 overexpression, including downregulation of SLC7A11 and GPX4 expression [Figure 3f-h] and upregulation of ACSL4 expression (P < 0.001, [Figure 3f and i]). These results indicate that PAQR3 promotes ferroptosis in HCC cells by enhancing lipid peroxidation, ROS and Fe2+ accumulation, and by regulating key ferroptosis-associated proteins.
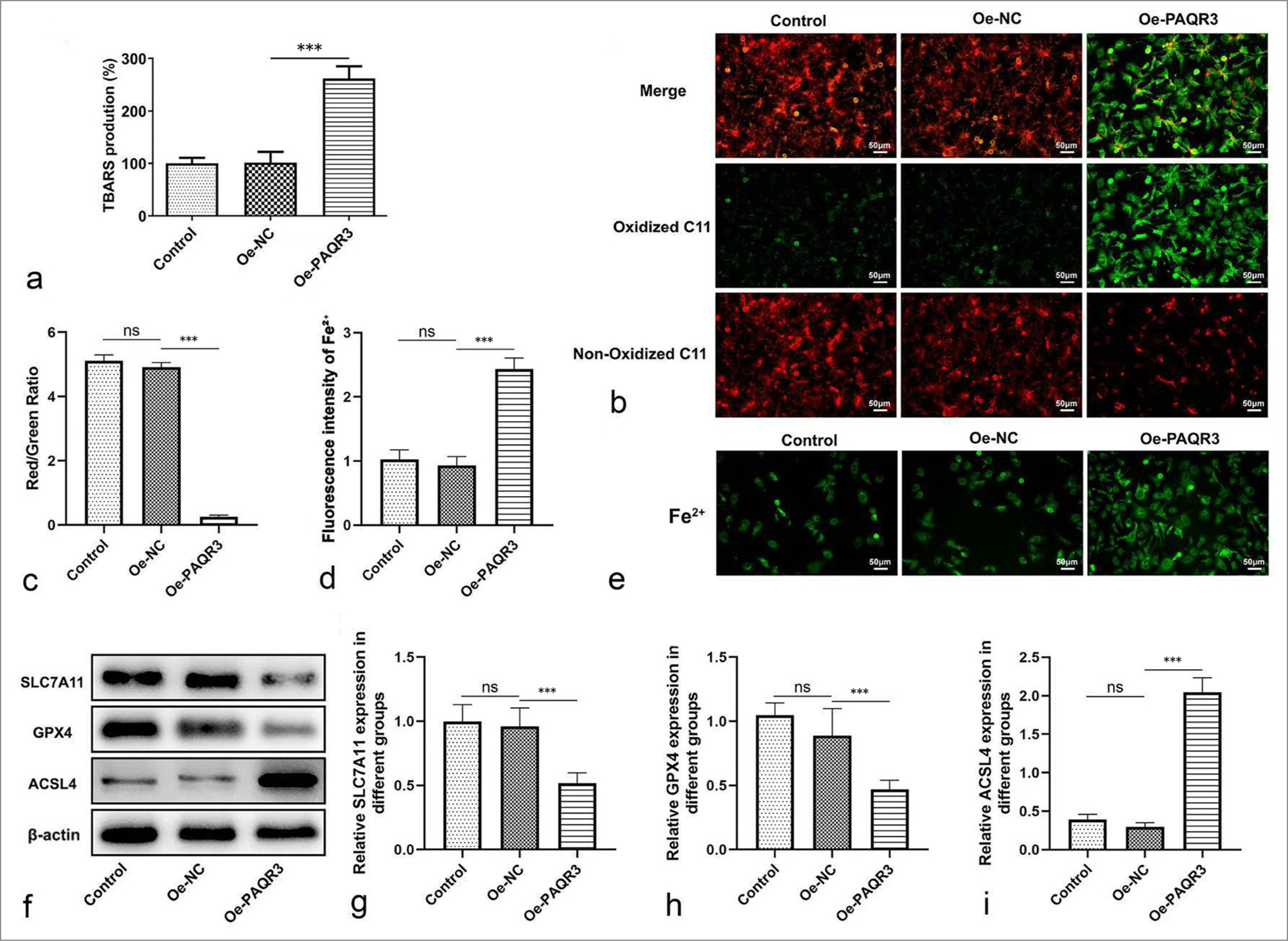
Export to PPT
Overexpression of PAQR3 inhibits the TGF-β pathwayTo further explore the effect of PAQR3 on the TGF-b pathway, the expression of key pathway-related proteins was examined. Western blot analysis showed that overexpression of PAQR3 led to a significant reduction in TGF-b1, p-Smad2, and p-Smad3 levels in HCC cells [Figure 4a]. Quantitative analysis further confirmed a marked decrease in TGF-b1 expression in the Oe-PAQR3 group compared with the control and Oe-NC groups (P < 0.001, [Figure 4b]). Similarly, p-Smad2 [Figure 4c] and p-Smad3 [Figure 4d] expression levels were markedly reduced upon PAQR3 overexpression (P < 0.001). Collectively, these findings indicate that PAQR3 overexpression suppresses the TGF-b/Smad signaling pathway in HCC cells.
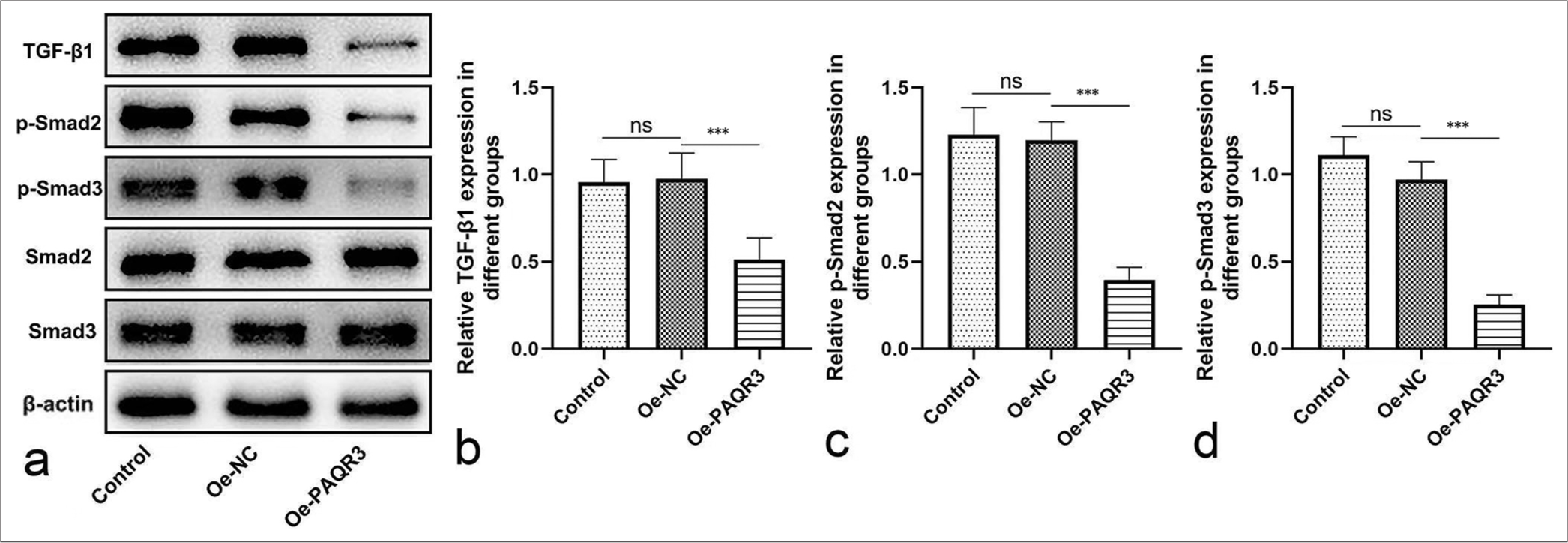
Export to PPT
PAQR3 regulates HCC cell metastasis and ferroptosis through the TGF-β pathwayTo investigate whether the effects of PAQR3 on HCC cells are mediated by the TGF-b pathway, cells were treated with TGF-b1 stimulation and the TGF-b inhibitor SB-431542. Wound healing and Transwell assays demonstrated that TGF-b1 treatment significantly promoted cell migration and invasion, whereas SB-431542 attenuated these abilities compared with the Oe-PAQR3 group (P < 0.001, [Figure 5a-d]). WB analysis further revealed that PAQR3 overexpression increased E-cadherin expression and decreased N-cadherin and Vimentin levels; these effects were partially reversed by TGF-b1 stimulation and further strengthened by SB-431542 treatment (P < 0.001, [Figure 5e-h]). Next, we examined ferroptosis-related changes. TBARS assays showed that lipid peroxidation levels were markedly elevated by PAQR3 overexpression, partially suppressed by TGF-b1, and enhanced by SB-431542 (P < 0.05, [Figure 5i]). Consistently, ROS accumulation, assessed by C11-BODIPY staining and red/green fluorescence ratio, followed the same trend. Similarly, intracellular Fe2+ levels were increased by PAQR3, reduced by TGF-b1, and further elevated by SB-431542 (ROS: P < 0.05; Fe2+: P < 0.001, [Figure 5j-m]). Western blot analysis of ferroptosis-related proteins showed that PAQR3 overexpression downregulated SLC7A11 and GPX4 [Figure 5n-p]) while upregulating ACSL4 [Figure 5q]; these changes were reversed by TGF-b1 and reinforced by SB-431542. Collectively, these results demonstrate that PAQR3 regulates both EMT-mediated metastasis and ferroptosis in HCC cells through the TGF-b/Smad signaling pathway.
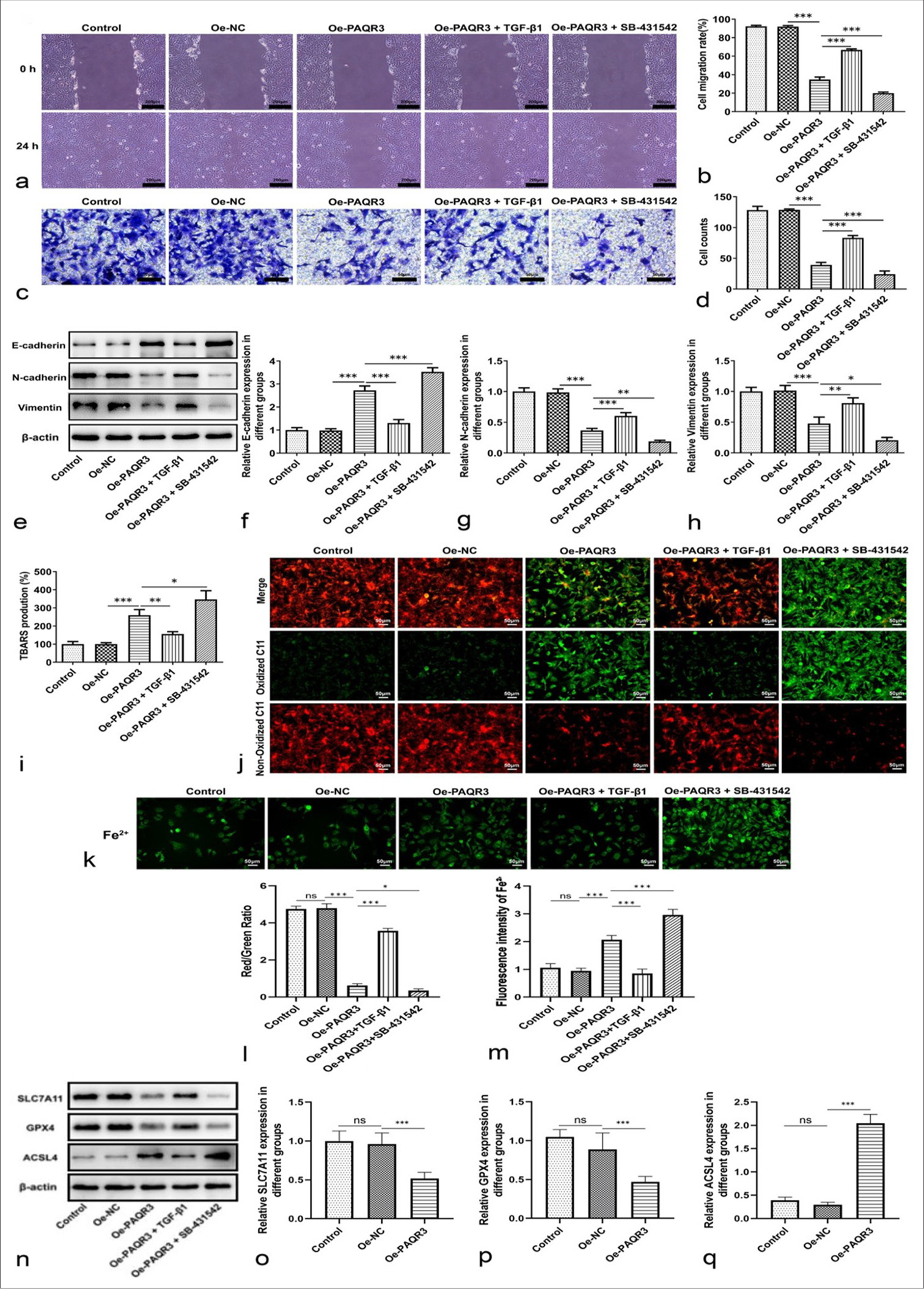
Export to PPT
DISCUSSIONPAQR3 has been reported to suppress cell migration, sprouting, proliferation, and angiogenesis, whereas its knockdown enhances these processes, highlighting a broad tumor-suppressive function in vitro.[15] In a study by Llovet et al., PAQR3-/- mice exhibited an increased number and size of papillomas, shortened tumor latency, and elevated keratinocyte proliferation.[16] Low PAQR3 expression has also been observed in colorectal cancer, gastric cancer, liver cancer, and other malignancies, where it correlates with tumor progression and poor prognosis.[17] In the present study, we examined the role of PAQR3 in HCC progression and found that PAQR3 was downregulated in HCC tissues and cells. Functionally, PAQR3 inhibited the proliferation, migration, and invasion of HCC cells in vitro. Moreover, PAQR3 suppressed TGF-b1/Smad signaling, and activation of TGF-b1 partially counteracted the inhibitory effects of PAQR3 overexpression on HCC metastasis in vitro.
During EMT, the expression of cell adhesion molecules (i.e., E-cadherin) decreases, while the cytokeratin cytoskeleton shifts toward a vimentin-based structure, and cells acquire mesenchymal morphology.[18] Through EMT, epithelial cells lose their epithelial characteristics and polarity, including junctions with the basement membrane, while gaining mesenchymal traits such as improved invasion and migration, resistance to apoptosis, and extracellular matrix degradation capabilities.[19] EMT is a primary biological process through which epithelial-derived malignant tumor cells obtain migratory and invasive capacities. It has been reported that PAQR3 and p53 synergistically regulate tumorigenesis and participate together in EMT, suggesting that p53 functions as a “checkpoint” controlling EMT, and only tumor cells lacking this checkpoint can effectively undergo EMT and contribute to tumor invasion and metastasis.[20] Consistent with these observations, in Hep3B cells, PAQR3 overexpression increased E-cadherin while decreasing vimentin and N-cadherin levels, indicating inhibition of EMT.
Previous studies have shown that PAQR3 inhibits the proliferation of acute lymphoblastic leukemia and promotes ferroptosis by regulating Nrf2 stability.[21] Accordingly, we explored the effect of overexpression of PAQR3 on ferroptosis in HCC cells. The results demonstrated that PAQR3 overexpression in HCC cells led to significant rises in Fe2+, ROS, and lipid peroxidation levels. Concurrently, the expression of ferroptosis-linked proteins (e.g., GPX4 and SLC7A11) was markedly reduced, whereas ACSL4 protein levels were significantly elevated. These findings indicate that PAQR3 promotes ferroptosis in HCC cells.
EMT, a critical process in tumor progression and metastasis, is regulated by multiple signaling pathways, including Notch, TGF-b/Smad, AKT/mTOR, and Wnt/b-catenin.[22] Among these, TGF-b plays a central role in controlling EMT and serves as a fundamental regulator of cell growth and differentiation. Recent studies have revealed that TGF-b influences cell differentiation, proliferation, and immune function, including inhibition of endothelial and epithelial cell growth, suppression of lymphocyte differentiation, and reduction of immunoactive cell proliferation.[22] While it can suppress tumor growth in certain contexts, TGF-b also promotes invasion, metastasis, and immune evasion in cancer.[9] In TGF-b/Smad signaling, TGF-b kinase phosphorylates the C-terminal residues of Smad2 and Smad3, which then form a complex with Smad4 and translocate to the nucleus to regulate downstream gene expression, thereby driving EMT. In this study, analysis of TGF-b/Smad-associated proteins revealed that PAQR3 overexpression inhibited this pathway.[23] Following TGF-b1 treatment, the effects of PAQR3 on HCC cell migration, invasion, and ferroptosis were partially compensated, whereas the TGF-b1 inhibitor SB-431542 exerted the opposite effects.
While our results demonstrate that PAQR3 overexpression concurrently induces ferroptosis and inhibits metastasis in HCC cells, the precise mechanistic link between these two phenotypes remains an important question for future investigation.[24] It is possible that these are parallel, independent outcomes of PAQR3-mediated TGF-b pathway regulation. However, we cannot rule out potential crosstalk; for instance, ferroptosis-induced lipid peroxidation and cellular damage could indirectly impair migratory and invasive capabilities, a hypothesis supported by emerging evidence in other cancers.[25] Conversely, the metabolic reprogramming associated with metastatic progression might alter cellular sensitivity to ferroptosis. Therefore, although our study identifies PAQR3 as a novel regulator of both ferroptosis and metastasis in HCC, whether a direct causal relationship exists between these processes requires further elucidation and represents a critical next step in understanding the multifaceted tumor-suppressive functions of PAQR3.[8,23]
This study has certain limitations that warrant consideration. First, the lack of detailed clinical information, including age, gender, and TNM stage, restricted our ability to perform stratified analyses and to adequately account for potential confounders. Second, the functional characterization of PAQR3 was primarily conducted in a single HCC cell line, and no in vivo evidence, such as xenograft mouse models, was performed. These factors may limit the generalizability of our findings and the strength of the therapeutic implications. Third, the regulatory role of PAQR3 in the TGF-b pathway requires further validation, particularly through direct binding assays to determine whether PAQR3 interacts with the TGF-b promoter or Smad response elements. Finally, the precise molecular mechanisms underlying PAQR3-mediated modulation of TGF-b signaling also remain to be elucidated. Future investigations incorporating comprehensive clinical datasets, diverse cellular models, and in vivo validation will be essential to validate and extend the present conclusions.
SUMMARYThis study demonstrates that PAQR3 expression is markedly reduced in HCC tissues and cells, and its overexpression suppresses HCC cell migration and invasion by restraining EMT, as evinced by increased E-cadherin and decreased Vimentin and N-cadherin levels. Mechanistically, PAQR3 promotes ferroptosis through increased Fe2+, ROS, and lipid peroxidation, along with GPX4 and SLC7A11 downregulation and ACSL4 upregulation. Furthermore, PAQR3 exerts its tumor-suppressive effects partly through inhibition of the TGF-b/Smad signaling pathway, as TGF-b1 activation was partially reversed, whereas SB-431542 enhanced, its anti-metastatic and pro-ferroptotic effects of PAQR3. Collectively, PAQR3 regulates metastasis and ferroptosis in HCC through the TGF-b pathway. These results highlight PAQR3 as a potential therapeutic target for inhibiting HCC progression through modulation of EMT, ferroptosis, and TGF-b pathway.
AVAILABILITY OF DATA AND MATERIALSAll relevant data and material of this study are available from the corresponding author on request.
ABBREVIATIONSPAQR3: Progestin and adipoQ receptor 3
HCC: Hepatocellular carcinoma
TGF-β: Transforming growth factor-β
WB: Western blotting
ROS: Reactive oxygen species
EMT: Epithelial-mesenchymal transition
GPX4: Glutathione peroxidase 4
SLC7A11: Solute carrier family 7 member 11
ACSL4: Acyl-coa synthetase long chain family member 4
IHC: Immunohistochemistry
IRS: Immunoreactivity score
FBS: Fetal bovine serum
STR: Short-tandem repeat
TBARS: Thiobarbituric acid reactive substance
BHA: Butylated hydroxyanisole
PGSK: Phen green SK
DAB: Diaminobenzidine
DMEM: Dulbecco’s modified eagle medium
RIPA: Radioimmunoprecipitation assay
SDS-PAGE: Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
PVDF: Polyvinylidene fluoride
HRP: Horseradish peroxidase
Oe-NC: Overexpression negative control
AUTHOR CONTRIBUTIONSXL: Conceived and designed the study, performed the experiments, and wrote the initial draft of the manuscript; YD: Responsible for data collection, performed the formal data analysis, and participated in the critical revision of the manuscript for important intellectual content. WL: Conducted the statistical analysis, validated the results, prepared the figures, and contributed to the reviewing and editing of the manuscript. All authors have reviewed the final manuscript and agree to be accountable for their own contributions. All authors are eligible for ICMJE authorship.
Comments (0)