Chemicals
Umbelliferone was acquired from Sigma-Aldrich Chemical (St. Louis, MO, USA). The following antibodies were obtained from different sources: Rabbit polyclonal antibody of ZO-1 (Cat# 61-7300) was supplied by Invitrogen (Carlsbad, CA, USA). Rabbit polyclonal antibody of Filaggrin (Cat# ENZ-ABS181) was supplied by Enzo Life Sciences (Farmingdale, NY, USA). Mouse monoclonal antibodies of Bcl-2 (Cat# ab692) and F-actin (Cat# ab205) were supplied by Abcam Biochemicals (Cambridge, UK). Rabbit polyclonal antibody of Bax (Cat# sc-20067) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse monoclonal antibodies of Occludin (Cat# sc-5562), Integrin β1 (Cat# sc-8978), RhoA (Cat# sc-418), ROCK (Cat# sc-17794), Rac1 (Cat# sc-514583), Cdc42 (Cat# sc-8401), AQP3 (Cat# sc-518001), HAS2 (Cat# sc-365263), and HYAL1 (Cat# sc-101340) were also obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse monoclonal β-actin antibody (Cat# A1978) was obtained from Sigma-Aldrich Chemical (St. Louis, MO, USA). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG and goat anti-mouse IgG were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA).
Cell Culture
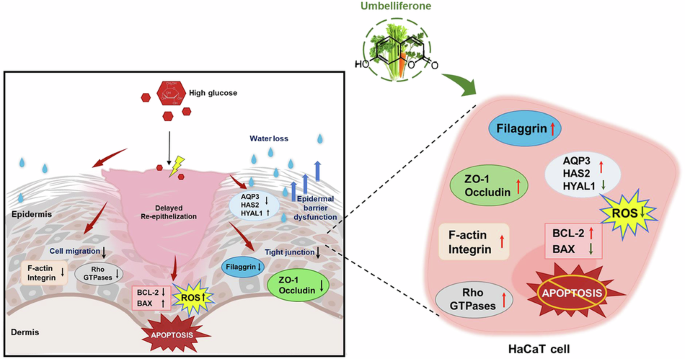
The immortalized human keratinocyte cell line (HaCaT) was obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM glutamine, 0.75 μg/ml human epidermal growth factor, and 75 μg/ml hydrocortisone at 37 °C humidified atmosphere of 5% CO2 in air. HaCaT were sub-cultured at 85% confluence. To mimic a hyperglycemia environment, immortalized keratinocytes were incubated in media containing 33 mM glucose, compared with normal media containing 5.5 mM glucose. Additionally, HaCaT were incubated for 3days in media containing 1–20 μM unbelliferone.
The viability of HaCaT was assessed using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-dephenyltertrazolium bromide) assay. The cells were incubated with 1 mg/ml MTT solution at 37 °C for 3 hours, resulting in the formation of an insoluble purple formazan product. This product was then dissolved in 250 μl of isopropanol. The optical density of the solution was measured using a microplate reader at a wavelength of 570 nm.
In vivo animal experiments
Adult male db/db mice (C57BLKS/+Leprdb Iar; Jackson Laboratory, Sacramento, CA, USA) and age-matched non-diabetic male db/m littermates (C57BLKS/J; Jackson Laboratory, Sacramento, CA, USA) were used in this study. Since db/db mice typically develop hyperglycemia by 7–8 weeks of age, 7-week-old mice were selected for the experiment. Mice were maintained under specific pathogen-free conditions on a 12-hour light/dark cycle at 23 ± 1 °C and 50 ± 10% relative humidity. They were provided with a standard laboratory chow diet (CJ Feed, Seoul, Republic of Korea) and had ad libitum access to water at the Hallym University animal facility. Before beginning the experimental interventions, all animals were allowed a one-week acclimatization period. Mice were then divided into three subgroups (n = 9–10 per group): a non-diabetic db/m control group and two db/db groups. One db/db group received a daily oral gavage of umbelliferone (10 mg/kg body weight), while the other group received daily water gavage for 10 weeks. Throughout the study, no mortality or signs of exhaustion were observed in any group
All experiments were approved by the Hallym University Committee on Animal Experimentation and conducted in accordance with the University’s Guidelines for the Care and Use of Laboratory Animals (hallym(2023-18)).
Western blot analysis
Western blot analysis was performed using whole cell lysates obtained from HaCaT (3.5 × 105 cells/dish) and skin tissue extracts from mouse skin. The lysates were prepared using a lysis buffer containing 1 M β-glycerophosphate, 1% β-mercaptoethanol, 0.5 M NaF, 0.1 M Na3VO4, and protease inhibitor cocktails. Equal amounts of protein from HaCaT and skin tissue lysates were loaded and separated by electrophoresis on 8–15% SDS-PAGE gels, and then transferred onto nitrocellulose membranes. To block nonspecific binding, the membranes were incubated for 3 hours with either 3% fatty acid-free BSA or 5% nonfat dry skim milk. Subsequently, the membranes were incubated overnight at 4 °C with primary antibodies specific to the target proteins. After washing the membranes with Tris-buffered saline-Tween 20 for 10 minutes, they were incubated with secondary antibodies conjugated to HRP (goat anti-rabbit IgG, goat anti-mouse IgG, or donkey anti-goat IgG) for 1 hour. The levels of each target protein were detected using an Immobilon Western chemiluminescent horseradish peroxidase substrate (Millipore Corporation, Burlington, MA, USA) and detected through chemiluminescence using a ChemiDoc (Cytiva). As comparative controls, incubation with a mouse monoclonal β-actin antibody was also carried out. Quantitative analysis of Western blot results was conducted using densitometry. Band intensities were measured with ImageJ software (NIH, Bethesda, MD, USA) and normalized to the corresponding β-actin bands. Relative expression levels were calculated and statistically analyzed.
Rhodamine phalloidine staining
HaCaTs (0.7 × 10^5 cells/slide) were cultured and exposed to 33 mM glucose in the presence or absence of 1-20 μM umbelliferone. The cells were fixed with 4% formaldehyde for 10 minutes and washed with phosphate-buffered saline (PBS). Following that, cells were treated with 10 units of the fluorescent dye rhodamine phalloidin and incubated for 20 minutes. Additionally, nuclear staining was performed using 1 μg/mL of 4’,6-diamidino-2-phenylindole (DAPI). Each slide was then mounted using VectaMount mounting medium from Vector Laboratories (Burlingame, CA, USA. Fluorescent images were captured using an Axiomager Optical fluorescence microscope (Nicon GmbH, Düsseldorf, Germany). The protein level of F-actin was quantified using an image analysis program provided by the microscope system.
Scratch wound assay
To assess the impact of glucose on the motility of immortalized keratinocytes and the potential effects of umbelliferone, an in vitro scratch wound assay was conducted. HaCaT cells were seeded in 12-well plates and incubated for 24 hours in media containing 10% fetal bovine serum (FBS). Upon reaching confluency, a horizontal scratch was made in each well using a pipette tip. Following the scratch, the cells were incubated for an additional 24 hours in culture media, either with or without 33 mM high glucose, and with or without 1–20 μM umbelliferone. Images of the scratch wounds were captured in 2–3 microscopic fields per well using the microscope system. The extent of keratinocyte migration was indicated by the scratched area. Quantification of the wound area was performed using ImageJ software (NIH, Bethesda, MD, USA)
Immunostaining
HaCaT cells (0.7 × 10^5 cells/slide) were fixed with 4% formaldehyde for 10 minutes, followed by permeabilization with 0.1% Triton X-100 on ice. To prevent nonspecific protein binding, cells were blocked with 5% bovine serum albumin (BSA) for 1 hour. The primary antibody for ZO-1 was diluted 1:100 in 5% BSA and incubated overnight at 4 °C. For cytochemical staining, FITC-conjugated anti-rabbit IgG (Sigma-Aldrich Chemical, St. Louis, MO, USA) was diluted 1:500 in PBS and applied for 30 minutes. The primary antibody for AQP3 was also diluted 1:200 in 5% BSA and incubated overnight at 4 °C, followed by staining with Cy3-conjugated anti-mouse IgG (Sigma-Aldrich Chemical, St. Louis, MO, USA) diluted 1:500 in PBS for 30 minutes. Nuclear staining was performed with 4’,6-diamidino-2-phenylindole (DAPI) diluted 1:1000 in PBS, and slides were mounted with glycerol (Sigma-Aldrich Chemical, St. Louis, MO, USA). Images were acquired using an inverted microscope (Nikon, Tokyo, Japan) for visualization.
ROS formation
Dihydroethidium (DHE, Invitrogen, Carlsbad, CA, USA) staining was performed to assess ROS production in HaCaT cells. HaCaT cells (0.7 × 10^5 cells/slide) were fixed with 4% formaldehyde for 10 minutes, followed by permeabilization with 0.1% Triton-X100 on ice for 10 minutes. Cells were then incubated with 20 μM DHE for 1 hour to stain for ROS. Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI) for 10 minutes, and stained cells were mounted with a mounting medium. Skin tissue specimens were also prepared by fixation and sectioned to 8 μm thickness, followed by DHE staining. Images of the stained slides and tissue sections were captured using an inverted optical microscope (Nikon, Tokyo, Japan).
Oxidative generation in HaCaT cells was assessed using 2’,7’-dichlorofluorescin diacetate (DCF-DA) staining. Cells challenged with 33 mM glucose were incubated with 10 μM DCF-DA in pre-warmed DMEM for 30 minutes at 37 °C. After dye loading, cells were washed thoroughly with phosphate-buffered saline, and nuclear staining with DAPI was performed. Images of the cultured cells were also captured using an optical microscope.
Hoechst 33258 staining
After incubating HaCaT cells for 15 minutes on glass-covered 24-well plates, cells were treated with 10 µg/mL Hoechst 33258 (Promega Co., Madison, WI, USA) for nuclear staining. Cells exhibiting fragmented or condensed nuclei were categorized as apoptotic. Each slide was imaged using an optical microscope system to detect nuclear morphology.
Immunohistochemical staining
For immunohistochemical staining, skin samples were collected at the end of the experiments and fixed in 10% buffered formalin. The paraffin-embedded skin tissues were sectioned at 8 μm thickness, deparaffinized, and rehydrated through xylene and graded ethanol solutions. Sections were incubated overnight with a primary antibody against ZO-1, followed by FITC-conjugated anti-rabbit IgG. Nuclear staining was performed with 4′,6-diamidino-2-phenylindole (DAPI, Santa Cruz Biotechnology, Santa Cruz, CA, USA). Each slide was mounted with VectaMount mounting medium (Vector Laboratories, Burlingame, CA, USA). Images were captured using an optical microscope.
Hematoxylin and eosin staining of skin tissue and measurement of epidermis/dermis thickness
Paraffin-embedded skin specimens were cut into 8 μM thick sections, dewaxed and rehydrated, and stained with eosin reagent to assess and hematoxylin for counterstaining. The stained tissue slides were examined using a microscope equipped. The thickness of skin epidermis and dermis was analyzed by ImageJ software (NIH, Bethesda, MD, USA).
Data analysis
Data were analyzed using SPSS (SPSS Inc., Chicago, IL, USA), with a significance level set at p < 0.05. Significant differences between treatment groups, indicated by different letters in the figures, were determined using one-way ANOVA. Tukey’s post hoc test was subsequently applied to identify specific group differences. Results are expressed as mean ± SD.

Comments (0)