
Remember me
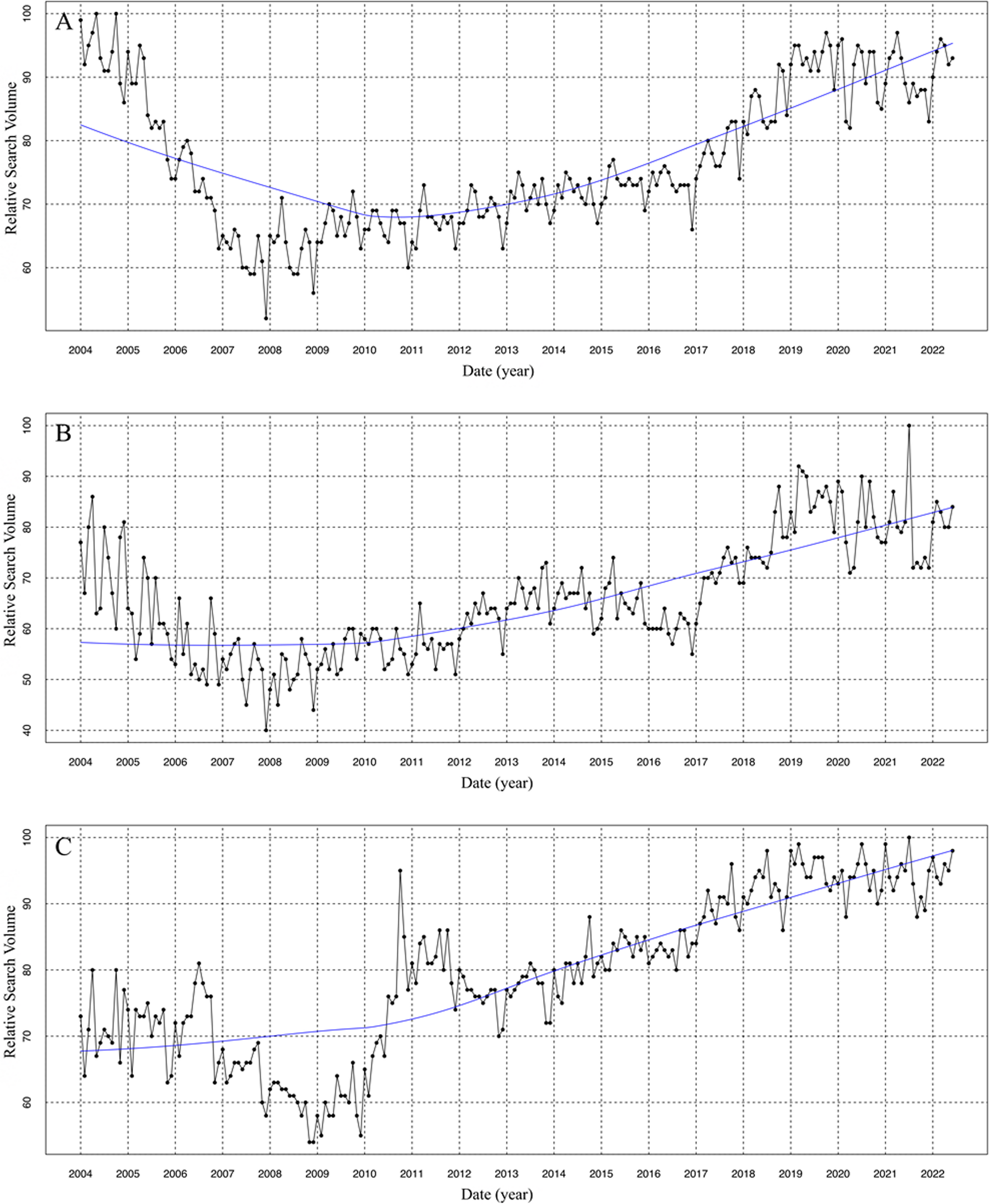
To construct an IgAN cell model, we treated HK2 cells with varying concentrations of IgA for 24 h. This treatment was designed to mimic the pathological conditions observed in IgAN, where abnormal IgA deposition plays a key role in kidney injury. By using different IgA concentrations, we aimed to identify the optimal concentration that induces significant cellular responses, such as inflammation or cell apoptosis. CCK-8 assay results proved that cell viability progressively decreased with increasing IgA concentrations (Fig. 1A). Furthermore, the content of pro-inflammatory factors, including IL-6, IL-1β, and TNF-α, were significantly increased in a dose-dependent manner (Fig. 1B). These findings suggest that IgA treatment induces an inflammatory response in HK2 cells, a hallmark feature of IgAN. To balance between reducing cell viability and maintaining cell survival for subsequent experiments, we selected an IgA concentration of 100 \(\mu\)g/mL for further studies.
Fig. 1
High expression levels of HRD1 are involved in IgAN progression. A CCK-8 assay was used to detect the cell viability after treatment with various concentrations of IgA. B ELISA kit was used to detect the content of IL-6, IL-1β, and TNF-α in HK2 cells after treatment with different concentrations of IgA. C Changes in the expression levels of HRD1 protein after treatment with different concentrations of IgA. D RT-PCR was employed to assess the interference efficiency after knocking down HRD1. E Changes in HRD1 protein expression levels after HRD1 down-regulation in HK2 cells. F Changes in cell viability after knockdown of HRD1 in HK2 cells. G Changes in cell apoptosis after HRD1 down-regulation in HK2 cells. H Changes in the levels of IL-6, IL-1β, and TNF-α after HRD1 knockdown in HK2 cells. *P < 0.05, **P < 0.01, ***P < 0.001
Next, western blot was performed to detect the expression levels of HRD1 protein in IgAN cell model. The results showed that the protein expression levels of HRD1 were gradually increased with increasing IgA concentrations (Fig. 1C). This suggests that HRD1 plays a critical role in the progression of IgAN.
To investigate the role of HRD1 in IgAN, we used shRNAs to decrease the expression levels of HRD1 in HK2 cells. RT-PCR was performed to select the optimal shRNAs for HRD1 knockdown efficiency, and the results showed that the sh-HRD1-1 exhibited the lowest levels of HRD1 mRNA (Fig. 1D). A similar reduction in HRD1 protein expression was observed after HRD1 down-regulation (Fig. 1E). Consequently, we selected this cell line for further investigation. CCK-8 assay results showed that the loss of HRD1 significantly enhanced cell viability (Fig. 1F). Flow cytometry analysis revealed a significant reduction in cell apoptosis after HRD1 knockdown (Fig. 1G). Additionally, the levels of pro-inflammatory factors, including IL-6, IL-1β, and TNF-α, were significantly decreased upon HRD1 down-regulation (Fig. 1H). Taken together, our findings demonstrated that the deficiency of HRD1 has a protective effect on IgAN injury, as it reduced cell apoptosis, enhanced cell viability, and decreased inflammatory response.
Inhibition of hrd1 alleviates renal injury in igan miceTo further verify the regulatory effects of HRD1 on IgAN in vivo, we used C57BL/6 mice to establish an IgAN mice model. Compared to the mice in sham groups, the mice in IgAN groups exhibited significantly increased HRD1 expression (Figs. 2A and B), along with enhanced IgA deposition in the kidneys (Figs. 2C). Histological analysis of the glomeruli from IgAN mice revealed compensatory expansion and epithelial cell swelling (Fig. 2D), collagen fiber deposition (Fig. 2E), and thickening of both glomerular and tubular basement membranes (Fig. 2F). Moreover, increased cell death was observed in the IgAN group (Figs. 2G). Additionally, the content of proteinuria, creatinine, and BUN (blood urea nitrogen) were significantly elevated (Figs. 2H), indicating renal dysfunction. Taken together, these data indicated that we have successfully established an IgAN mice model, which recapitulates the key pathological features of IgAN observed in humans, including kidney injury, glomerular changes, and increased renal dysfunction.
Fig. 2
Down-regulation of HRD1 alleviates renal injury in IgAN. A Changes in HRD1 expression levels after HRD1 down-regulation in IgAN mice. B Changes in HRD1 protein expression levels after HRD1 down-regulation in IgAN mice. C Changes in IgA expression levels after HRD1 knockdown in IgAN mice. D HE staining was used to detect the pathological changes in renal tissues. E Masson staining was employed to evaluate collagen fiber deposition. F PAS staining was used to detect the lesions in glomeruli and tubules. G TUNEL was used to assess cell apoptosis after knockdown of HRD1 in IgAN mice. H Changes in the levels of proteinuria, creatinine and BUN in IgAN mice with HRD1 deficiency. The image above is taken at 200 × magnification, and the image below is taken at 400 × magnification (Figs. 2D-F). *P < 0.05, **P < 0.01, ***P < 0.001. Scale bars: 50 μm (A, C, D, E, and F)
The lentivirus was used to downregulate HRD1 expression in IgAN mice to investigate the effect of HRD1 deficiency on IgAN. Compared to the IgAN + sh-NC group, the IgAN + sh-HRD1 group showed significantly decreased HRD1 expression (Figs. 2A and B), and reduced IgA deposition (Figs. 2C). Histological analysis of the glomeruli in the IgAN + sh-HRD1 group revealed improved glomerular structure (Fig. 2D), decreased collagen fiber deposition (Fig. 2E), and reduced thickening of both glomerular and tubular basement membranes (Fig. 2F). Additionally, there was a notable reduction in cell death (Fig. 2G). Furthermore, the levels of proteinuria, creatinine, and BUN, were significantly decreased in IgAN + sh-HRD1 group (Figs. 2H). Taken together, these findings highlight the potential of HRD1 as a therapeutic target for mitigating IgAN progression and provide a useful in vivo model for further investigations.
Identification of mfn1 as a function target of hrd1To investigate the mechanism by which HRD1 contributes to IgAN, we used the Ubi Browser website to search for the potential target proteins of HRD1. Based on this analysis, we selected KIF1B, MLLT4, and MFN1 for further research (Fig. 3A). The results of RT-PCR and western blot analysis revealed that only the protein levels of MFN1 were significantly reduced or increased upon HRD1 up-regulation or down-regulation (Figs. 3B and C). In contrast, the levels of KIF1B and MLLT4 mRNA and protein expression showed no significant changes (Figs. 3B and C). Therefore, we selected MFN1 as a potential key target in HRD1-mediated IgAN progression.
Fig. 3
HRD1 promotes the ubiquitination of MFN1 protein. A Using Ubi Browser website to predict HRD1 interactions protein. B Changes in the expression levels of KIF1B, MLLT4 and MFN1 mRNA after HRD1 up-regulation or down-regulation. C Changes in the expression levels of KIF1B, MLLT4 and MFN1 protein after HRD1 up-regulation or down-regulation. D Changes in MFN1 protein expression levels after knockdown of HRD1 in HK2 cells. E: Changes in MFN1 protein expression levels after down-regulation of HRD1 in IgAN mice. F Changes in the levels of MFN1 ubiquitination after HRD1 down-regulation. F Western blot was used to assess the protein stability of MFN1 after HRD1 down-regulation. *P < 0.05, **P < 0.01, ***P < 0.001
We next examined the expression levels of MFN1 in IgAN. Western blot results showed that the levels of MFN1 expression were reduced both in HK2 cells and in IgAN mice (Figs. 3D and E). Whereas, the loss of HRD1 led to an increase in MFN1 expression (Figs. 3D and E). These data suggested that HRD1 is located upstream of MFN1 and inhibits its expression, and this regulation is important for the pathogenesis of IgAN.
To further investigate the mechanism by which HRD1 downregulates MFN1 expression, we considered previous studies that showed HRD1 promotes the ubiquitination of MFN1 [27]. Based on these findings, we hypothesized that HRD1 regulates the ubiquitination of MFN1 in HK2 cells. To test this, we transfected HK2 cells with HA-Ub, shRNAs targeting HRD1 expression, and myc-MFN1 vectors to assess the ubiquitination of MFN1. As shown in Fig. 3E, the levels of MFN1 ubiquitination were elevated in the presence of HRD1, while the loss of HRD1 inhibited MFN1 ubiquitination. Furthermore, the protein stability of MFN1 was decreased upon HRD1 expression, whereas the loss of HRD1 led to enhanced protein stability of MFN1 (Fig. 3F). In conclusion, our findings strongly suggested that HRD1 contributes to IgAN progression by promoting the ubiquitination of MFN1, thereby reducing the protein stability of MFN1.
Mfn1 down-regulation accelerates igan progressionAs MFN1 acts as a suppressor gene in the progression of IgAN, we next investigated whether over-expression of MFN1 could inhibit IgAN progression. To do this, we over-expressed MFN1 in HK2 cells and IgAN mice. The results of western blot analysis showed that the expression levels of MFN1 were significantly increased after MFN1 up-regulation (Fig. 4B). Additionally, cell viability was increased (Fig. 4C), and cell apoptosis was reduced (Fig. 4E) in HK2 cells after MFN1 over-expression. Moreover, over-expression of MFN1 led to a decrease in the levels of IL-6, IL-1β, and TNF-α (Fig. 4D). These data indicated that MFN1 over-expression enhances the survival of HK2 cells, which may contribute to inhibiting the progression of IgAN.
Fig. 4
HRD1 promotes IgAN progression by downregulating MFN1 expression. A RT-PCR was employed to assess the interference efficiency after knocking down MFN1. B Changes in MFN1 protein expression levels after HRD1 and MFN1 knockdown in HK2 cells. C Changes in cell viability after HRD1 and MFN1 knockdown in HK2 cells. D Changes in the content of IL-6, IL-1β, and TNF-α after knocking down HRD1 and MFN1 in HK2 cells. E Changes in cell apoptosis after HRD1 and MFN1 knockdown in HK2 cells. F Changes in MFN1 expression levels after HRD1 and MFN1 down-regulation in IgAN mice. G Changes in MFN1 protein expression levels after HRD1 and MFN1 down-regulation in IgAN mice. H Changes in IgA expression levels after HRD1 and MFN1 down-regulation in IgAN mice. I HE staining was used to detect the pathological changes after HRD1 and MFN1 down-regulation in IgAN mice. J PAS staining was used to detect the lesions in glomeruli and tubules after HRD1 and MFN1 down-regulation in IgAN mice. K Masson staining was employed to assess collagen fiber deposition after HRD1 and MFN1 down-regulation in IgAN mice. L Changes in cell death after HRD1 and MFN1 down-regulation in IgAN mice. M Changes in the levels of IL-6, IL-1β, and TNF-α after HRD1 and MFN1 down-regulation in IgAN mice. *P < 0.05, **P < 0.01, ***P < 0.001. Scale bars: 50 μm (F, H, I, J, K and L)
To further confirm whether MFN1 upregulation also inhibits IgAN progression in vivo, we examined the effects of MFN1 overexpression in IgAN mice. As shown in Figs. 4F and G, the expression levels of MFN1 were significantly enhanced after MFN1 overexpression. More importantly, over-expression of MFN1 led to a significant reduction in IgA deposition (Fig. 4H). Additionally, MFN1 over-expression improved glomerular and tubular lesions (Figs. 4I and J), inhibited collagen fiber deposition (Fig. 4K), reduced cell death (Fig. 4L), and decreased the levels of proteinuria, creatinine, and BNU (Fig. 4M). These results strongly suggest that MFN1 up-regulation attenuates the malignant progression of IgAN by reducing inflammatory response, renal injury, and fibrosis.
Since the down-regulation of HRD1 significantly alleviates IgAN injury (Fig. 1 and Fig. 2) and HRD1 downregulates MFN1 expression, we next examined whether the loss of MFN1 would accelerate IgAN progression. We used RT-PCR to screen for the optimal knockdown efficiency and found that the sh-MFN1-2 exhibited the lowest expression of MFN1 mRNA (Fig. 4A). In accordance with these results, the protein levels of MFN1 were also significantly reduced (Fig. 4B). Thus, we selected this cell line for our next experiments.
Notably, the loss of MFN1 exacerbates the renal injury that had been alleviated by HRD1 downregulation. In vitro, the deficiency of MFN1 markedly decreased cell viability (Fig. 4C), increased levels of inflammatory factors (Fig. 4D), and increased cell death (Fig. 4E). In vivo, MFN1 downregulation led to increased IgA deposition (Fig. 4H), more severe glomerular and tubular lesions (Figs. 4I and J), increased collagen fiber deposition (Fig. 4K), elevated cell apoptosis (Fig. 4L), and worsened kidney dysfunction (Fig. 4M). These findings strongly demonstrated that HRD1 contributes to IgAN progression by inducing MFN1 ubiquitination, highlighting the pivotal role of MFN1 in the pathogenesis of IgAN.
Hrd1 induces mitochondrial dysfunction through mfn1As MFN1 is a critical factor for mitochondrial fusion [8, 10, 28], we hypothesized that mitochondrial function may be impaired in IgAN. To test our hypothesis, we used MitoSOX to assess the levels of ROS (reactive oxygen species) in mitochondria, JC-1 to detect the mitochondrial membrane potential, Mito-Tracker to label mitochondria. The results of MitoSOX staining showed that the levels of ROS in mitochondria were significantly increased in both HK2 cells and IgAN mice (Figs. 5A and C), indicating mitochondrial oxidative stress. JC-1 staining showed that the mitochondrial membrane potential was markedly decreased in HK2 cells (Fig. 5B), suggesting mitochondrial dysfunction. Additionally, Mitotracker staining showed a reduction in fluorescence intensity in IgAN mice (Fig. 5D), further supporting the loss of mitochondrial integrity. Furthermore, there was a reduction in ATPase activity in both HK2 cells and IgAN mice (Figs. 5E and F), which is consistent with the impairment of mitochondrial function, as APTase is essential for maintaining cellular energy homeostasis. Taken together, these data strongly indicated that mitochondrial dysfunction occurs during the progression of IgAN.
Fig. 5
HRD1 maintains mitochondrial function through MFN1. A Changes in mitochondrial ROS levels after HRD1 and MFN1 knockdown in HK2 cells. B Changes in mitochondrial membrane potential after knockdown of HRD1 and MFN1 in HK2 cells. C Changes in mitochondria after HRD1 and MFN1 down-regulation in IgAN mice. D Changes in mitochondrial ROS levels after down-regulation of HRD1 and MFN1 in IgAN mice. E Changes in ATPase activity in HK2 cells. F Changes in ATPase activity in IgAN mice. *P < 0.05, **P < 0.01, ***P < 0.001. Scale bars: 100 μm (A and B), 50 μm (C and D)
As MFN1 acts as a suppressor gene to inhibit IgAN progression, we next investigated whether MFN1 over-expression could restore mitochondrial function. We found that MFN1 overexpression obviously enhanced mitochondrial function, as evidenced by a reduction in ROS levels (Figs. 5A and C), indicating a decrease in mitochondrial oxidative stress. Additionally, mitochondrial membrane potential was significantly increased (Fig. 5B), indicating improved mitochondrial integrity. Moreover, Mitotracker fluorescence intensity was markedly enhanced (Fig. 5D). Furthermore, MFN1 overexpression led to a significant increase in ATPase activity (Figs. 5E and F), which further support the restoration of mitochondrial function. In summary, our data revealed that MFN1 plays a crucial role in preserving mitochondrial health, and its over-expression could potentially serve as a therapeutic strategy to mitigate mitochondrial dysfunction in IgAN.
As MFN1 is a down-stream factor of HRD1, we next tested whether the loss of MFN1 would accelerate mitochondrial damage. The results showed that the loss of HRD1 significantly improved mitochondrial function, while the loss of MFN1 reversed these benefits, as indicated by increased ROS levels (Figs. 5A and C), decreased mitochondrial membrane potential (Fig. 5B), and decreased Mitotracker fluorescence intensity (Fig. 5D), and reduced ATPase activity (Figs. 5E and F). In conclusion, our findings demonstrated that HRD1 leads to mitochondrial dysfunction by facilitating MFN1 ubiquitination.
Myc regulates the transcription level of hrd1To further investigate the upstream regulators of HRD1, we used the animal TFDB website to predict the transcription factors that can regulate the expression of HRD1. Among the predicted factors, we focused on myc, as the transcription levels of myc mRNA were increased in IgAN [25], indicating that myc plays a role in the progression of IgAN, while the underlying mechanism remains unclear. We selected three potential binding sites to examine whether myc could bind to HRD1 promoter region. The results of CHIP showed that site2 is a functional binding site for myc (Fig. 6A). We mutant the sequence of site 2 to assess the regulatory effect of myc on HRD1 transcription. The results from the dual luciferase system showed that the wild type form of HRD1 exhibited significantly higher transcription activity upon myc over-expressing, while the mutant form of HRD1 showed no transcription activity (Fig. 6B). These data indicated that myc binds to site 2 of the HRD1 promoter region, and this interaction is essential for promoting HRD1 transcription.
Fig. 6
Myc participates in IgAN progression by controlling HRD1 transcription levels. A CHIP assay was performed to detect the binding between myc and HRD1 promoter. B Dual luciferase reporter system was employed to detect the regulatory effect of myc on the HRD1 promoter. C Changes in myc protein expression levels in HK2 cells. D Changes in myc protein expression levels in IgAN mice. E RT-PCR was employed to assess the interference efficiency after knocking down myc. F Changes in expression levels of myc, HRD1, and MFN1 protein in HK2 cells. G Changes in cell viability in HK2 cells. H Changes in cell death in HK2 cells. I Changes in the content of IL-6, IL-1β, and TNF-α in HK2 cells. J Changes in ATPase activity in HK2 cells. K Changes in mitochondrial ROS levels in HK2 cells. L Changes in mitochondrial membrane potential in HK2 cells. *P < 0.05, **P < 0.01, ***P < 0.001. Scale bars: 100 μm (K and L)
To detect the levels of myc expression in IgAN, we performed western blot analysis. The results indicated myc expression was significantly increased both in HK2 cells and in IgAN mice (Figs. 6C and D), suggesting that myc is upregulated in the context of IgAN and is likely involved in IgAN progression.
To examine the role of myc in the progression of IgAN, we used shRNAs to knockdown myc expression in HK2 cells. RT-PCR results showed the sh-myc-2 exhibited the lowest expression of myc mRNA (Fig. 6E). Therefore, we selected this cell line for subsequent experiments. Western blot analysis revealed that the expression levels of myc and HRD1 were decreased, while the levels of MFN1 were increased (Fig. 6F), which is consistent with the notion that myc promotes HRD1 expression, while inhibiting MFN1 expression. CCK-8 assay indicated that the loss of myc increased cell viability (Fig. 6G), while cell apoptosis was decreased after myc knockdown (Fig. 6H). The content of pro-inflammatory factors, such as IL-6, and IL-1β, and TNF-α, were reduced after knockdown of myc (Fig. 6I). These findings indicated the deficiency of myc hindered IgAN progression by reducing inflammation and promoting cell survival.
Moreover, we investigated whether myc down-regulation could improve mitochondria function. The results revealed that enhanced ATPase activity (Fig. 6J), suggesting improved mitochondrial function. The results of Mito SOX and JC-1 staining showed that a reduction in ROS levels (Fig. 6K) and an increase in mitochondrial membrane potential (Fig. 6L). Taken together, these results indicated that myc deficiency attenuates IgAN injury by promoting cell survival and improving mitochondrial function. This highlights the potential of targeting myc as a therapeutic strategy for IgAN.
As HRD1 is a downstream factor of myc, we next examined whether HRD1 over-expression could reverse the effects of myc down-regulation. CCK-8 assay results showed that cell viability decreased after HRD1 up-regulation (Fig. 6G). Flow cytometry results revealed a significant increase in cell apoptosis after HRD1 overexpression (Fig. 6H). Furthermore, ELISA results revealed an increase in the content of IL-6, and IL-1β, and TNF-α (Fig. 6I). Additionally, ATPase activity was reduced (Fig. 6J). MitoSOX and JC-1 staining results further showed that ROS levels were increased (Fig. 6K) and mitochondrial membrane potential was diminished (Fig. 6L). Taken together, these findings strongly proved that myc led to mitochondrial dysfunction in IgAN progression by decreasing MFN1 expression through HRD1.
Comments (0)