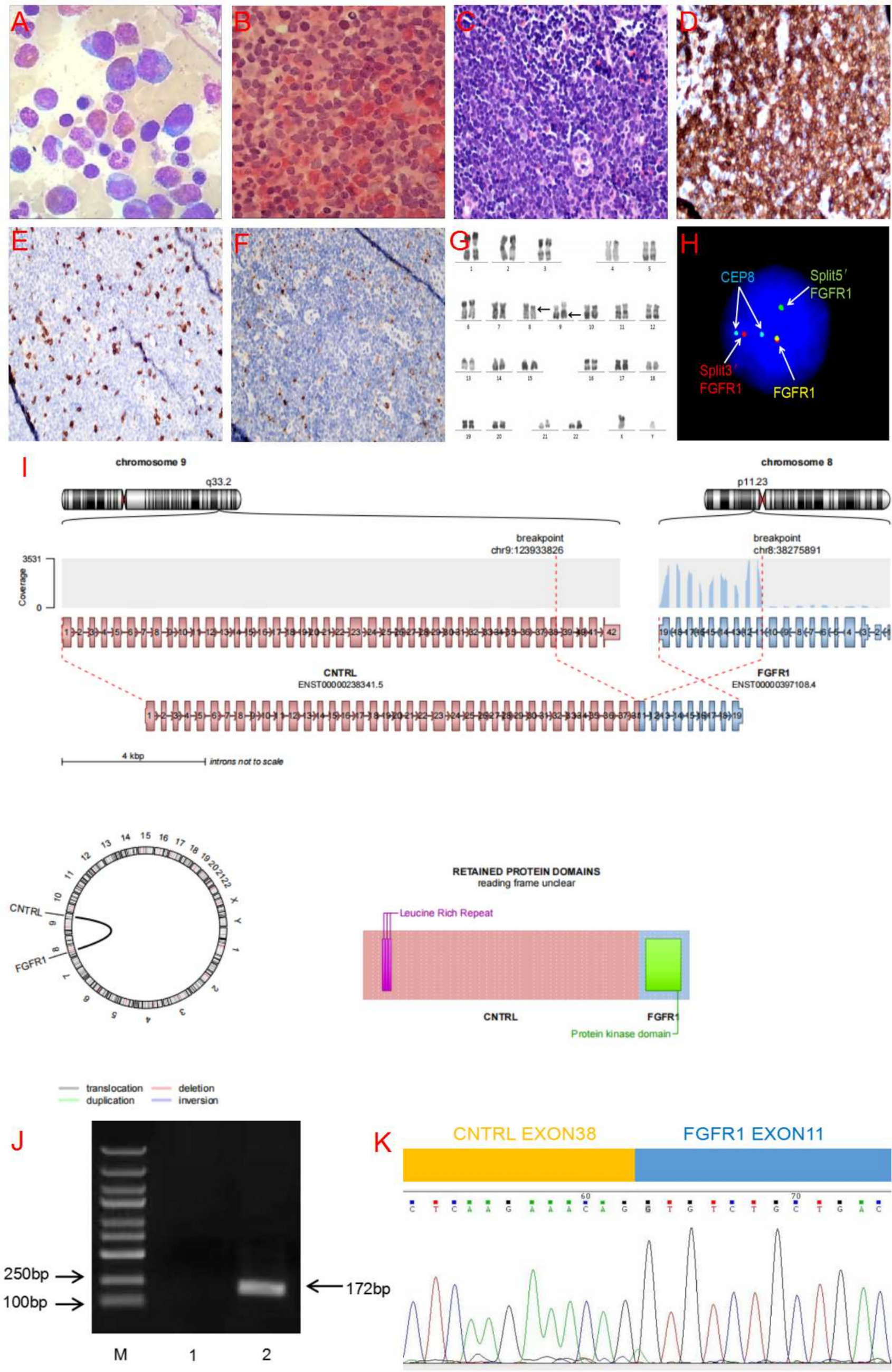
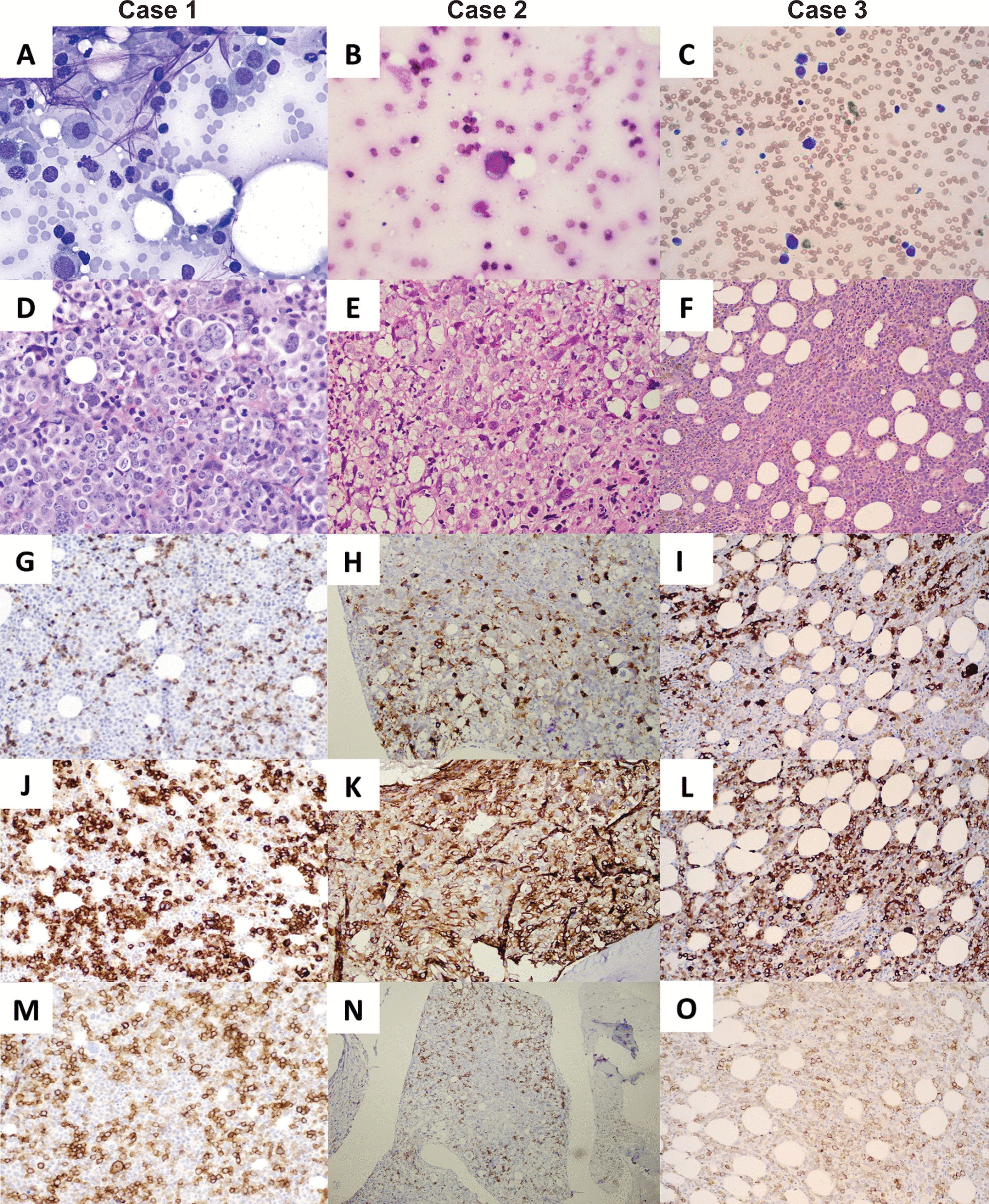
The 2022 ICC recognizes a category of AML with mutated TP53, which is defined by a TP53 mutation with a VAF ≥ 10%. The diagnosis of AML with TP53 mutation takes precedence over AML with myelodysplasia-related cytogenetic anomalies (AML-MRC) due to the poor prognosis TP53 mutations convey [10, 11]. The majority of identified cases of AML-EMD fall into AML with mutated TP53 (when mutational analysis was performed) or AML-MRC, with the exception of two cases occurring in patients with Down syndrome and Schwachman-Diamond syndrome respectively [3, 6].
In contrast, AML with TP53 mutations is not an independent classification in the WHO-HAEM5 [11]. Given their complex karyotypes, the majority of reported AML-EMD fall into the category of AML, myelodysplasia-related (AML-MR). The classification of AML-EMD secondary to germline predisposition follows a similar pattern as the ICC. For cases that fail to meet criteria for genetically defined AML, including those with insufficient material for testing as in patient 2 of this study, the WHO-HAEM5 recommends classification based on differentiation. However, a category to include overlapping erythroid and megakaryocytic differentiation does not currently exist.
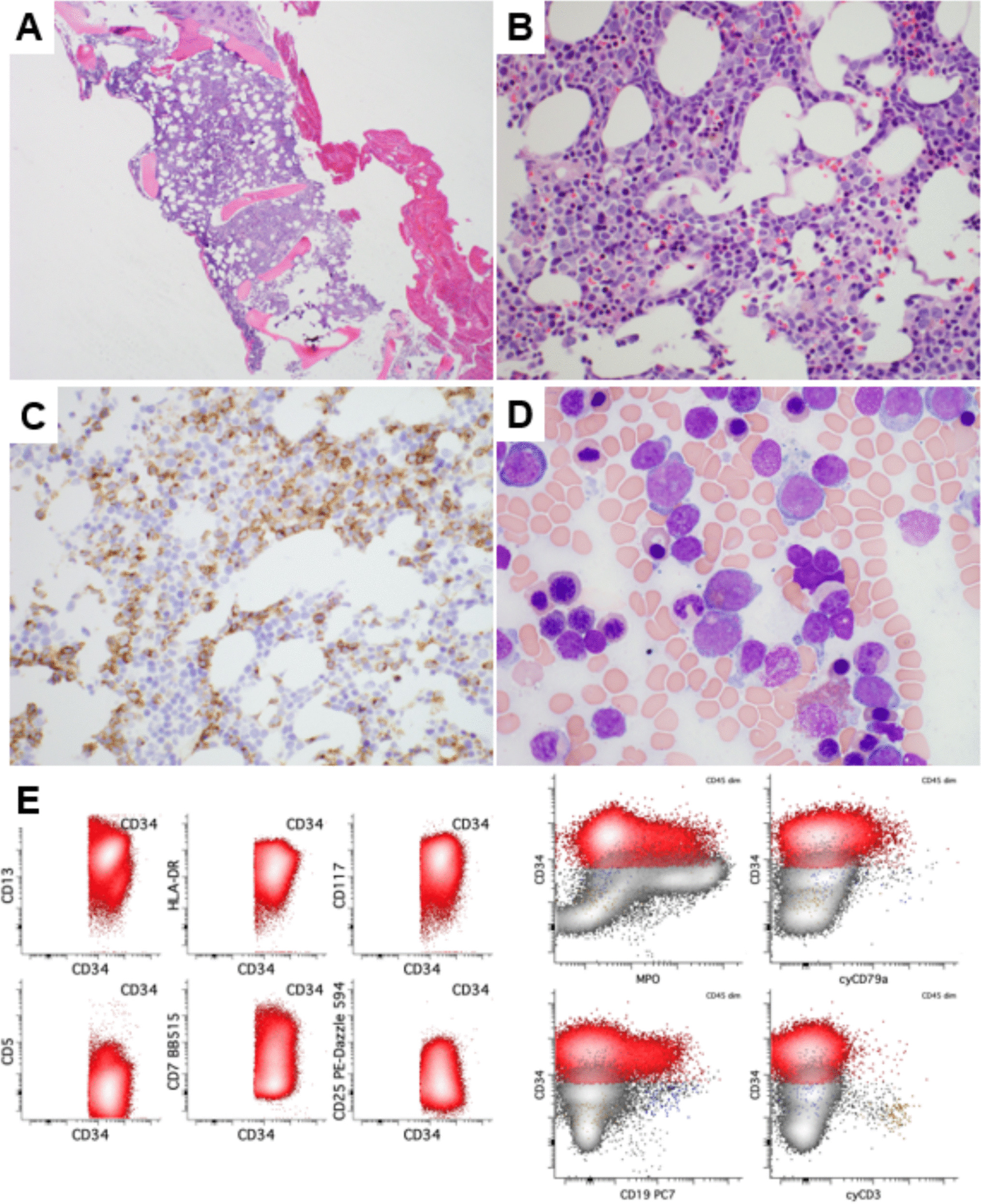
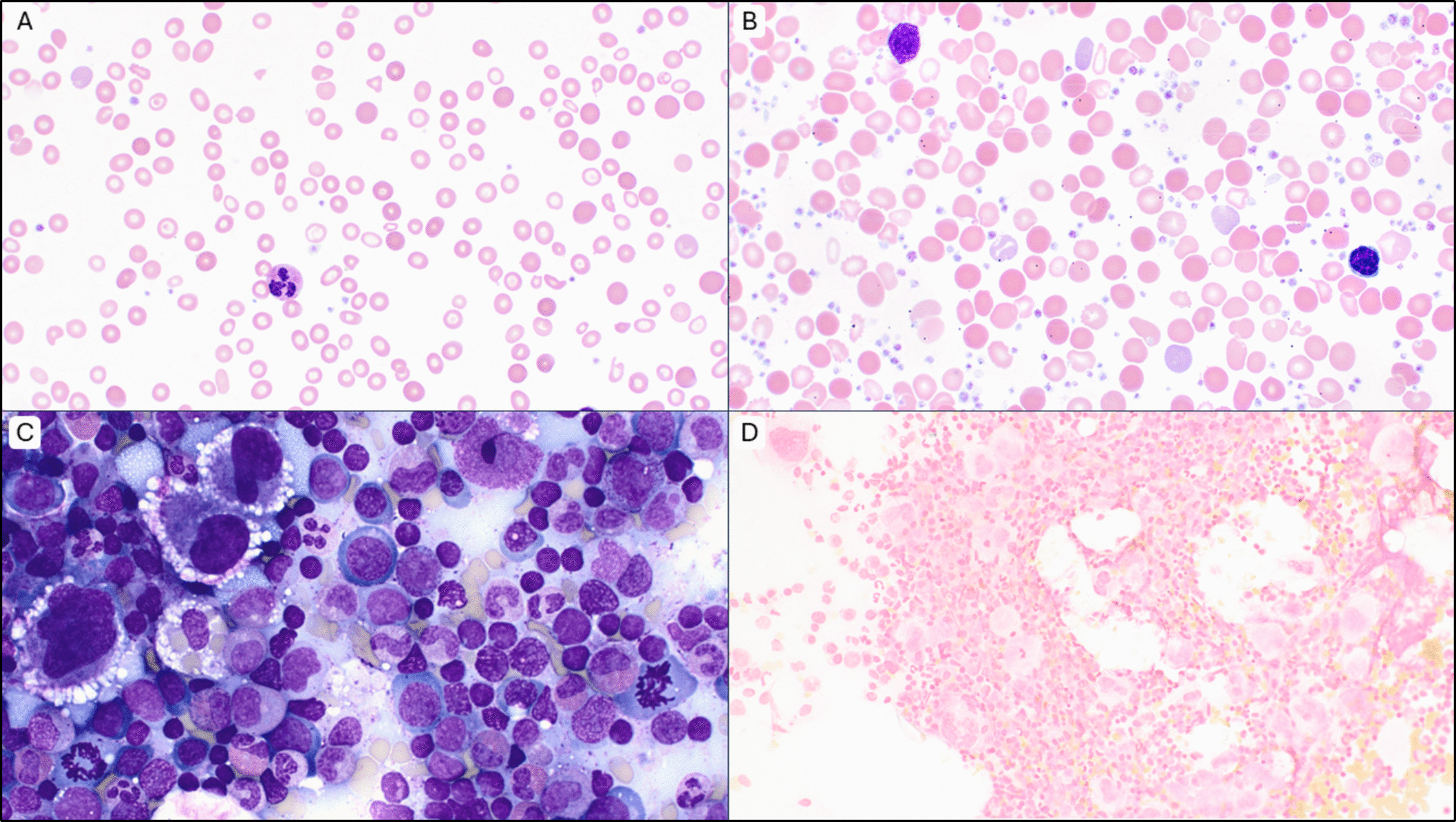
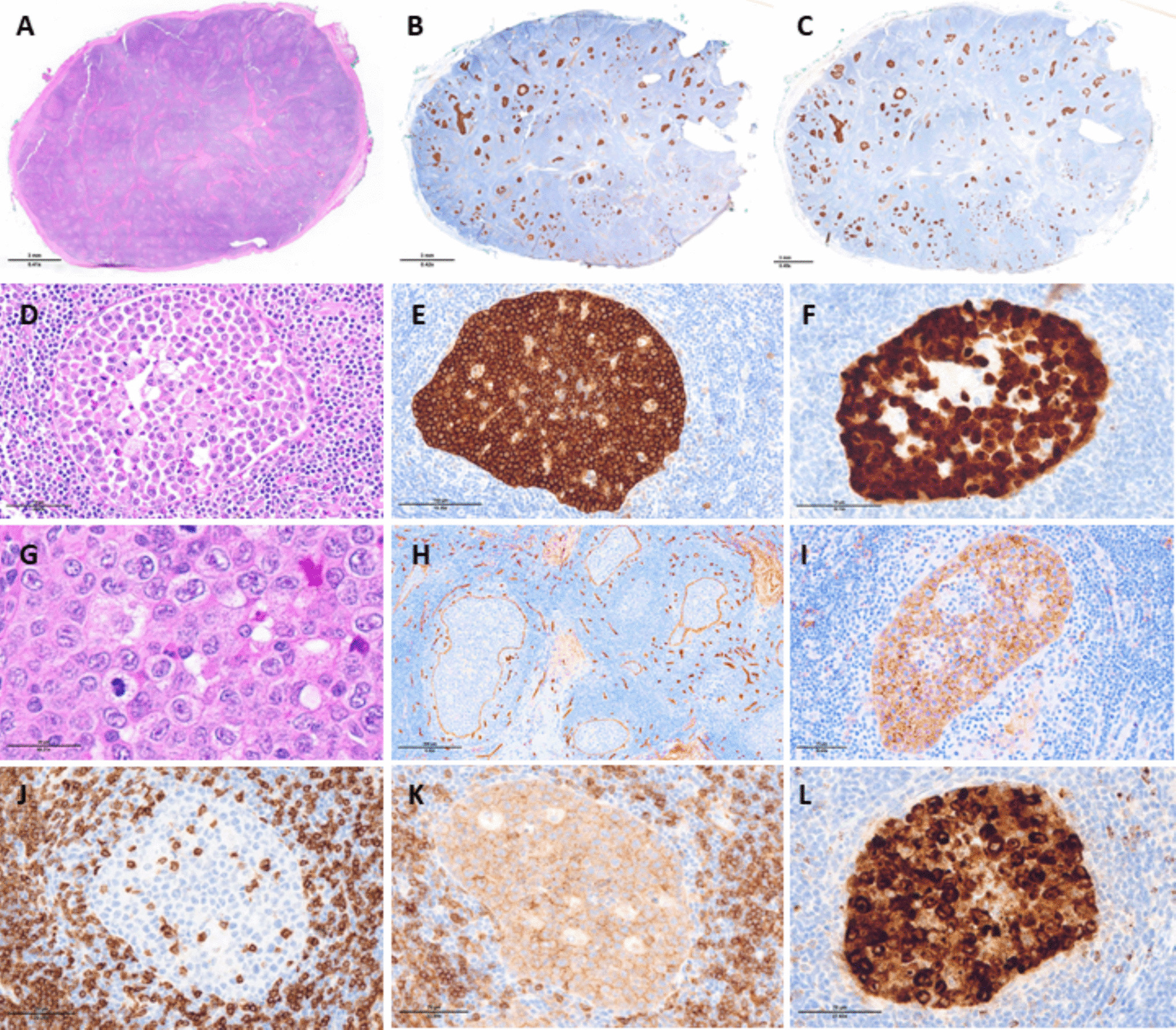
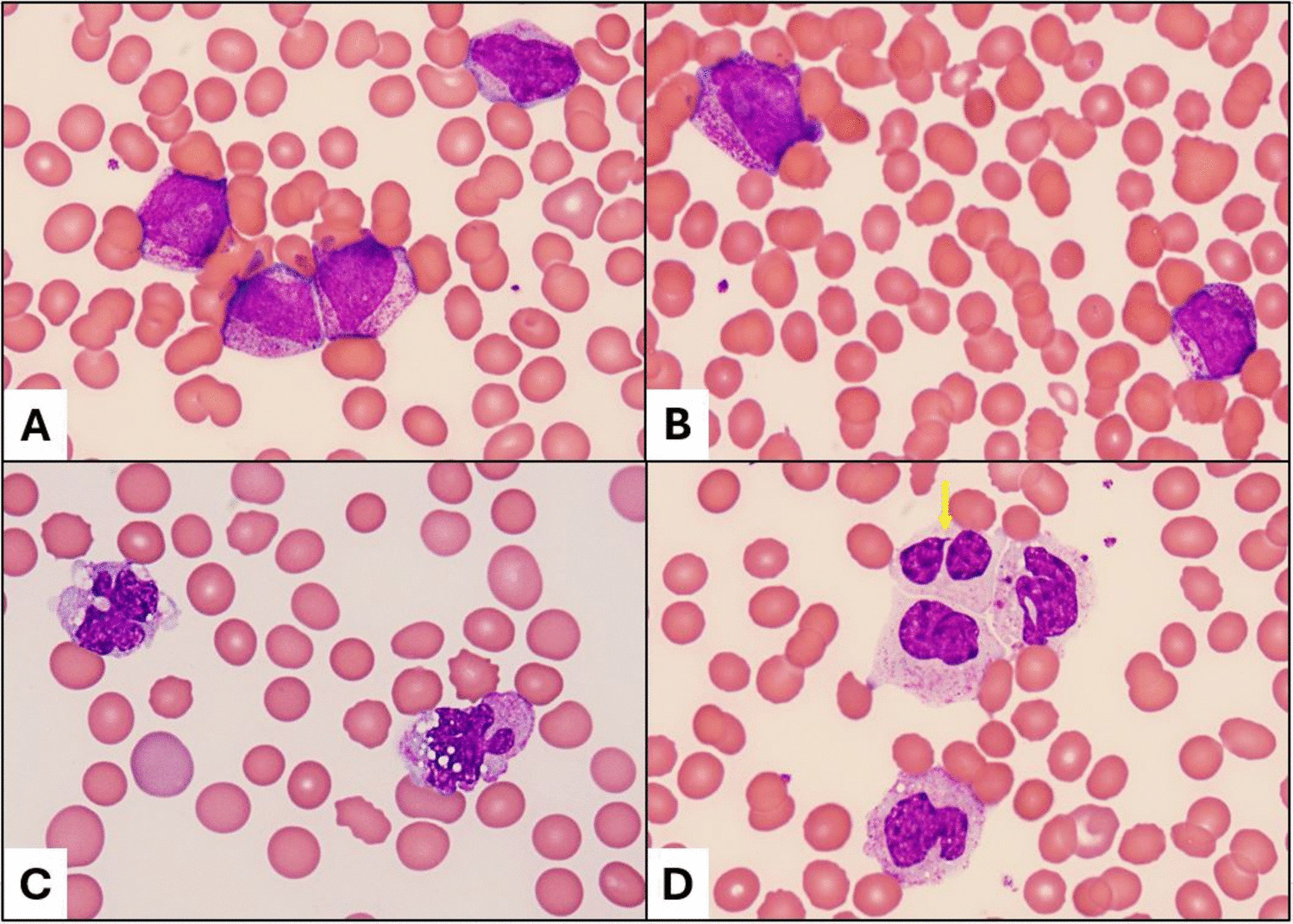
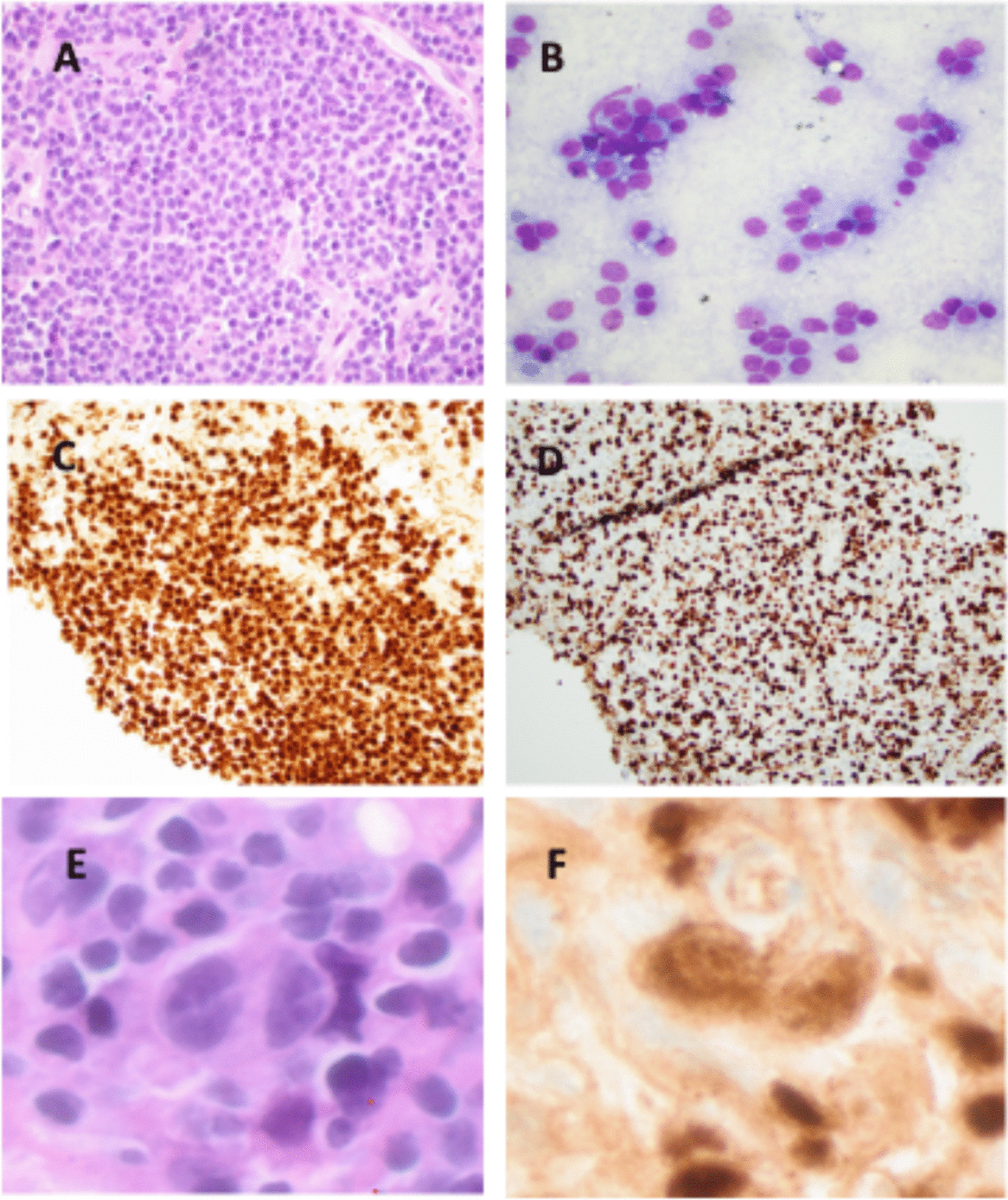
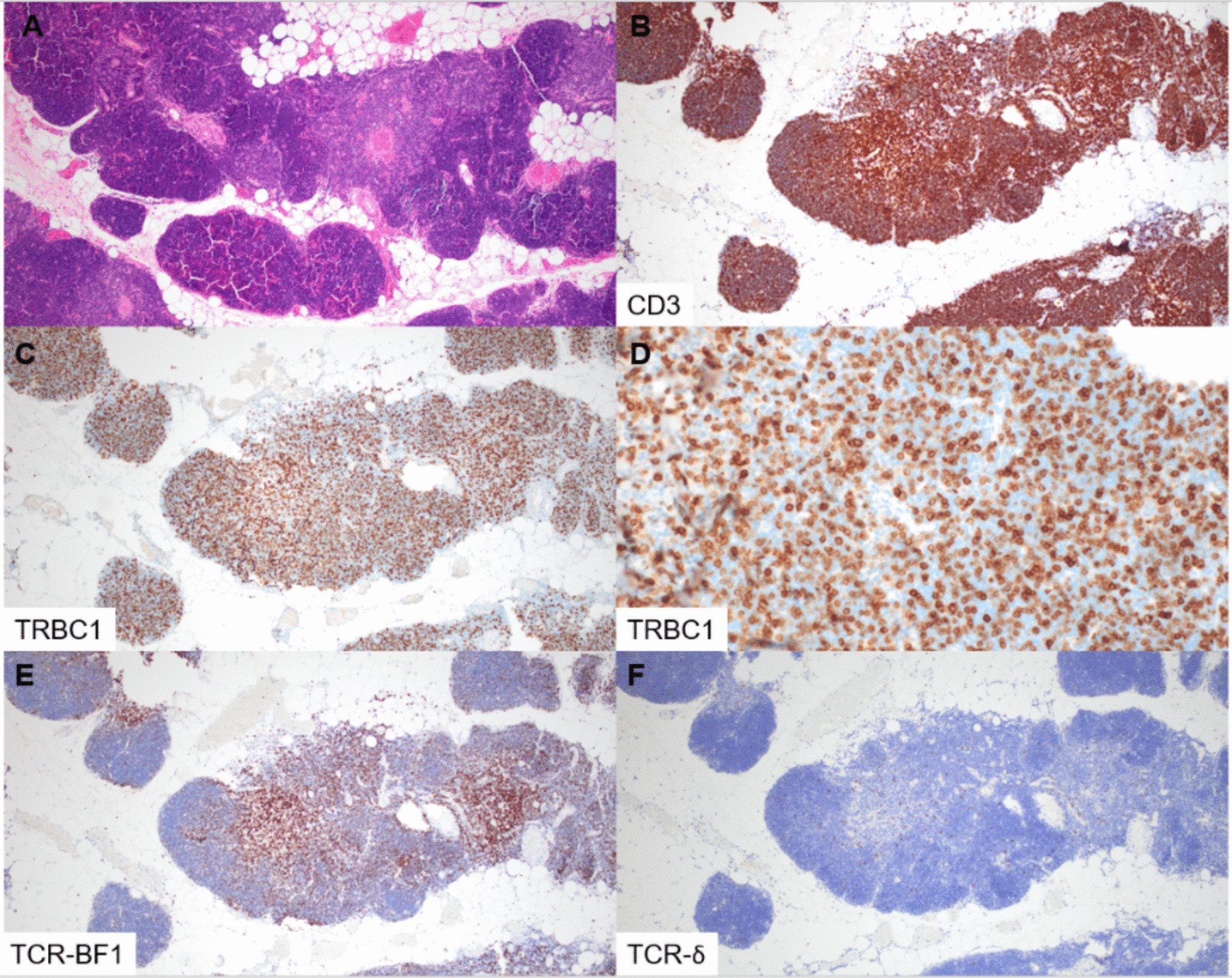
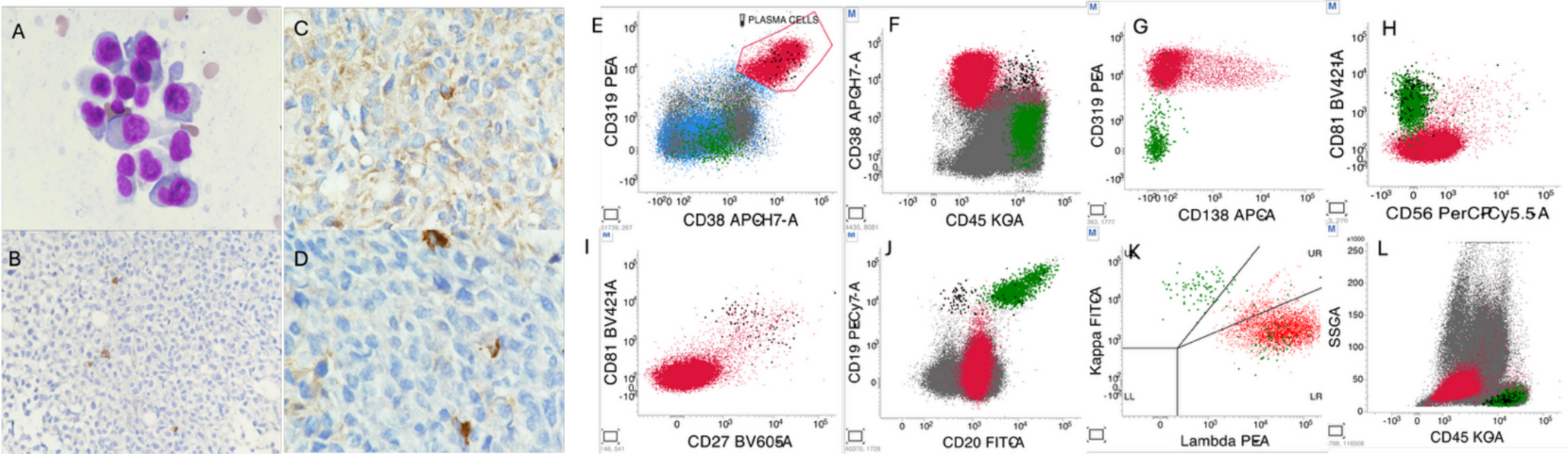
Overall, the genetic landscape of AML-EMD is complex. As mentioned, TP53 mutations are particularly enriched in these cases (6/7 tested patients) [4, 9]. AML-EMD with TP53 mutations appear to follow the trend of poor outcomes in other AMLs with TP53 mutations [11]. However, this is unlikely to be the only contributing factor as the dismal outcome was also observed in AML-EMD without identified TP53 mutations or alterations to chromosome 17p13.1. JAK1/2 mutations are similarly enriched in AML-EMD. In patient 2, prominent megakaryocytic hyperplasia and dysplasia hinted at the possibility of evolution from an underlying JAK2-mutated myeloproliferative neoplasm (MPN). Interestingly, we only observed JAK1/2 mutations in cases with concomitant TP53 mutations (3/6 patients). We also note a case of Down syndrome–associated AML-EMD that highlights an alternative GATA1-driven pathogenesis of dual erythroid and megakaryocytic differentiation [6, 12, 13].
Mechanistically, synergistic Jak2 V617F and loss-of-function Trp53 mutations have been noted to drive fully penetrant, transplantable AML in a murine model, pointing to a likely mechanism of transformation seen in many post-MPN AML and AML-EMD patients, involving BMP2/SMAD pathway activation [14, 15]. Jak2 and Trp53 biallelic inactivation together lead to an expansion of aberrant megakaryocytic-erythroid progenitors (MEPs), consistent with bipotential lineage markers seen in the identified cases in this study. A recent computational tool mapping AML patient samples onto a single-cell reference atlas of human hematopoiesis has also shed light on distinct differentiation states of genetically defined AML [16]. Recurrent genetic alterations in AML-EMD, including complex karyotypes and pathogenic mutations in TP53, JAK2, and GATA1 mutations found in AML-EMD, were associated with early erythroid and MEP/megakaryocytic progenitor differentiation states.
Importantly, identification of this unique differentiation state has relevance to clinical management. Similar to that described in TP53-mutated AML, erythroid/megakaryocytic differentiation may confer resistance to the BCL-2 inhibitor, venetoclax, through BCL-XL overexpression [17,18,19,20]. Recognizing that many of these cases harbor JAK2 mutations, combined BCL-XL/JAK inhibition may be a possible therapeutic avenue [19]. Given the significant biologic and prognostic overlap with TP53-mutated AML, a diagnosis of MDS/AML-EMD should certainly prompt immediate testing for TP53 loss-of-function mutations. If testing is not able to be performed, we suggest that pathologic diagnosis should note the presence of dual erythroid and megakaryocytic differentiation. With the exception of Down syndrome–related cases, clinical consideration may be given to treating presumptively as a TP53-like AML. In the future, updated classification frameworks may allow for more unambiguous terminology to define these rare cases.
Overall, these findings suggest that cases of reported AML-EMD likely represent biologically similar diseases, with diagnostic challenges and incongruent classifications by current WHO and ICC standards. Further studies are needed to examine leukemic transformation of AML-EMD lacking JAK2 and TP53 mutations. A deeper understanding of both the genetic and differentiation states of AML-MRD will complement existing strategies to improve diagnostic classification, risk stratification, and drug response prediction.

Comments (0)