Study design and objective
This study was designed as a prospective multicenter observational phase II trial evaluating the feasibility and safety of online adaptive MR-guided SBRT in cardiac/epicardial/paracardial lesions. Participating study centers comprise the radiation oncology departments of the University Hospital Heidelberg, the University Hospital Munich, the Agostino Gemelli University Hospital Rome, and the University Hospital Zurich. The primary endpoint is a combined endpoint for which occurrence of any of the following events is considered an event:
any acute toxicity > CTCAE grade 2 within 3 months after RT initiation (Common Terminology Criteria for Adverse Events CTCAE v5).
treatment discontinuation, whereby a connection with the treatment must exist.
mortality within 3 months after RT start (related to treatment and/or disease).
The secondary endpoint comprises the oncologic outcome, including local control and survival as well as acute and late toxicity. Furthermore, patient-reported outcome measures as well as the technical feasibility of treatment (intrafractional image quality and gating, frequency of plan adaptation, motion analysis, duty cycle of respiratory gating, physical assessment of treatment plans) represent further secondary study outcomes.
The study was set to start recruitment in 2022, with the aim of recruiting 10–15 patients per year due to the rare incidence. The inclusion period was planned to last 2 years but was then extended until the fourth quarter of 2025 by an amendment in 2024.
Patient population
Inclusion criteria according to the protocol are:
ability to comply with the study protocol and provide informed consent
age ≥ 18 years
inoperable primary or recurrent malignant cardiac sarcomas or cardiac/pericardial metastases or patients who refuse surgery
ECOG performance status 0–2
Exclusion criteria according to the protocol are:
patients without legal capacity or who are unable to understand the nature, significance, and consequences of the study
simultaneous participation in other interventional trials which could interfere with this trial
pregnancy
contraindication to MRI
Statistical analysis
A per-protocol analysis will be performed for all patients treated in the study. All results will be interpreted descriptively. Kaplan–Meier curves will be used to assess local control and overall survival rates. Adverse events will be reported with severity grades according to CTCAE. All data will be analyzed using SPSS Statistics as well as Excel (newest versions available; IBM Corp., Armonk, NY, USA and Microsoft, Redmond, WA, USA, respectively). Data management is performed using RedCap hosted at the IBE Munich (Institut für medizinische Informationsverarbeitung, Biometrie und Epidemiologie University Hospital LMU Munich).
Treatment planning: target delineation, planning objectives, and mRgSBRT delivery
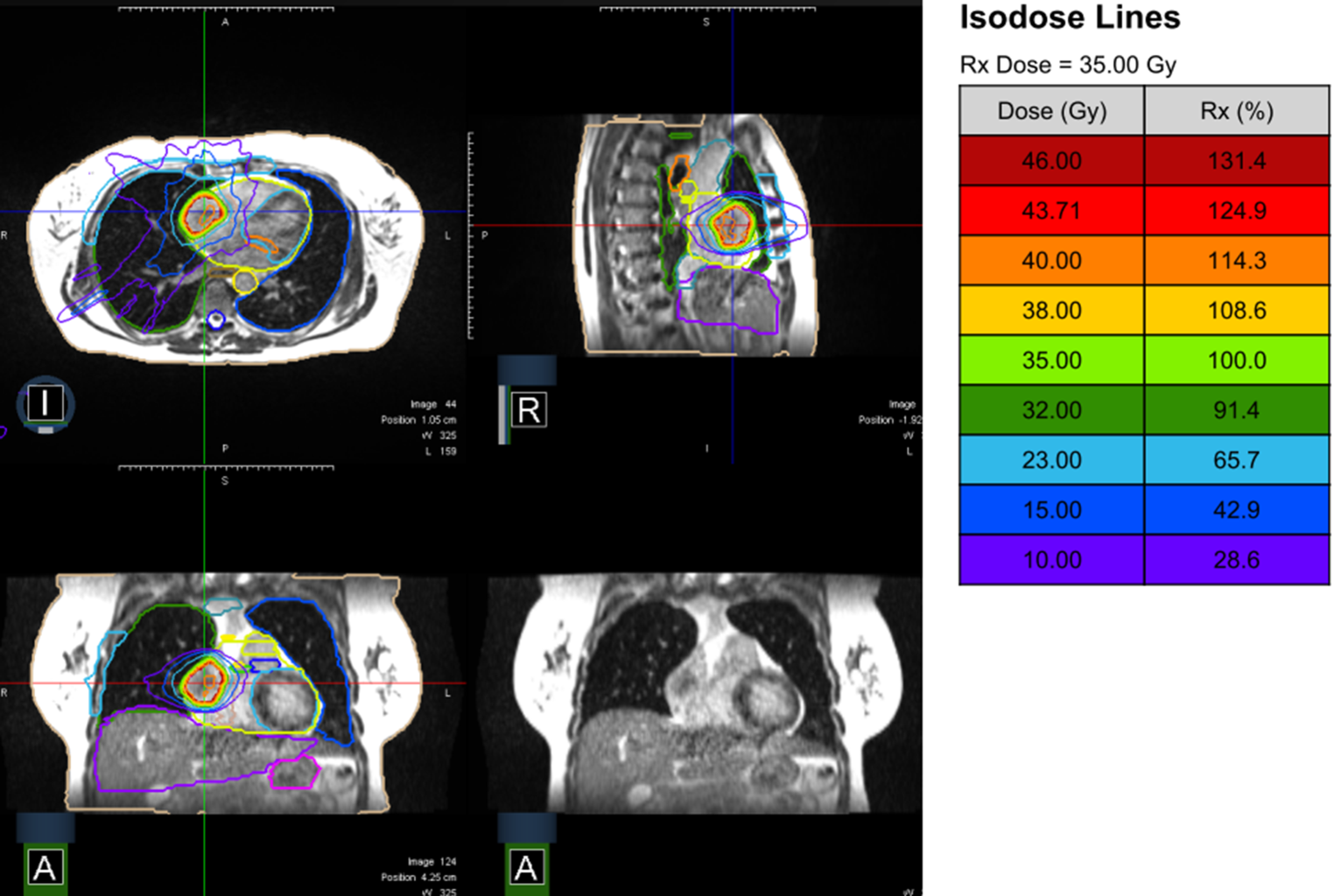
Radiotherapy planning follows the standard procedures of MR-guided RT of each institution and includes MRI simulation using a dedicated 0.35 T hybrid MR-Linac (ViewRay, Oakwood Village, OH, USA). Multiple scans using breath-hold techniques are usually acquired to obtain a reproducible and stable breath-hold and define the position best tolerated by the patient. Thereafter, a planning CT using the same patient positioning and the same breath-hold is performed for electron density information. These two imaging sets are then fused, and target delineation is performed. The target volume (gross tumor volume, GTV) and the organs at risk (OAR; heart, heart valves, heart minus PTV, aorta, spinal cord, spinal canal, lungs, trachea, esophagus, etc.) are delineated. The GTV is isotropically expanded by 3(–5) mm to define the planning target volume (PTV). The dose range is 5 × 6–8 Gy to the 80% isodose (non-consecutive days), depending on the tumor size and localization, and might be individualized on a case-by-case basis. During dose delivery, a uniform 3–5 mm margin is used for real-time gating, allowing a pixel excursion tolerance of 3%–8%. Gating is preferably performed on the GTV or, alternatively, by using a surrogate (Table 1).
Table 1 Dose constraints for treatment planning for 5 fractionsEligible patients who consent to participate in the study will undergo treatment as indicated and approved by an interdisciplinary tumor board.
Follow-up
Overall documentation at baseline and follow-up is shown in Table 2. Serial assessments before and every 3 months after radiation treatment will be performed.
Table 2 Workflow of the SHARP-studyAt baseline, documentation includes obtaining informed consent, recording demographic data and medical history, and conducting a clinical examination that encompasses the New York Heart Association (NYHA) classification as well as measurements of body weight and height. The ECOG performance status is assessed, along with documentation of current medication, particularly anticoagulation therapy. If possible, health-related quality of life (HR-QoL) will be assessed by the European Organization for Research and Treatment of Cancer (EORTC) Quality of Life Core Questionnaire for Cancer Patients (QLQ C30), the Kansas City Cardiomyopathy Questionnaire (KCCQ), and the New York Heart Association (NYHA) functional classification to classify the extent of heart failure. Diagnostic imaging via CT and/or MRI is performed. If possible, cardiac function is assessed via transthoracic echocardiography, including detailed evaluation of left ventricular parameters such as Simpson biplane measurements, 3D left ventricular ejection fraction, 3D systolic and diastolic volumes, global longitudinal strain, and diastolic function. Right ventricular parameters are also measured, including TAPSE (tricuspid annular plane systolic excursion), RV diameters, RV systolic and diastolic volumes, 3D RV function, and global longitudinal strain. Both left and right atrial dimensions are documented, along with an evaluation of the heart valves. A laboratory examination (creatinine, troponin, creatine kinase [CK], CK-MB, NT-proBNP) can be additionally performed.
At the end of radiation treatment, the feasibility of the MRgSBRT treatment is assessed, as are treatment discontinuation, interruptions, and surpassed dose constraints. Clinical examination is performed, and medication changes and acute toxicity during radiation treatment are registered. If possible, the quality of life assessment is repeated.
At subsequent study visits (every 3 months), documentation continues with clinical examinations that include the NYHA score, body weight, and height measurements. The ECOG performance status and medication records are updated. If possible, cardiac function is again assessed using transthoracic echocardiography with the same detailed parameters for the left and right ventricles, atria, and valve evaluations as at baseline. If possible, laboratory examinations can be performed using the same parameters as for baseline. Quality of life assessments using the QLQ-C30 and KCCQ are repeated. Additionally, documentation at these visits includes recording acute and late toxicities according to the Common Terminology Criteria for Adverse Events (CTCAE) v5.0 as well as survival status. Regular imaging with CT and/or MRI will be performed for the evaluation of local disease control, as defined in the institutional standard operating procedures (SOPs).
Study coordination and registration
The study is coordinated by the Department of Radiation Oncology at the LMU Munich, with the principal investigator based at the LMU. The LMU Munich oversees trial management, database administration, and reporting. Patient recruitment and treatment are carried out at the radiation oncology departments of the University Hospitals of Heidelberg, Munich, Zurich, and the Agostino Gemelli University Hospital in Rome.
The study is conducted in accordance with the Declaration of Helsinki and has received ethics approval from the Ethics Committee of the Medical Faculty of the LMU Munich (no. 21-0696) and is registered at the German Study Registry (no. DRKS00027108). The study is also registered as an ARO study (study number 2024-09). Informed consent is obtained from all participants.

Comments (0)