
Remember me

All organisms that store triacylglycerols (TGs) rely on lipolysis to release fatty acids and glycerol for energy use and to regulate lipid distribution between tissues. In simpler organisms, lipid droplets are present across many cell types and lipolysis operates locally within individual cells [1]. As some multicellular organisms increased in size and complexity and adapted to repeated feeding–fasting cycles, adipose tissue emerged as a specialized organ that centralizes lipid storage and release. In this role, adipose tissue buffers fluctuations in nutrient availability, protects non-adipose tissues from ectopic lipid accumulation, and supports systemic metabolic stability [2]. Beyond energy storage, adipose tissue functions as a dynamic endocrine organ whose regulation influences the integrity of multiple tissues and overall systemic metabolic health. Disruption of adipocyte function, particularly lipolysis, is therefore associated with a wide range of pathological states [3].
Dysregulated lipolysis is observed across diverse metabolic conditions, including obesity, diabetes, rare genetic disorders, and inflammatory or immune-mediated diseases. In diabetes and metabolically unhealthy obesity, defective suppression of lipolysis in the fed state and inadequate stimulation during fasting impair the temporal control of fatty acid flux, contributing to insulin resistance and broader features of metabolic syndrome [4]. Rare disorders such as congenital generalized and partial lipodystrophy further illustrate the consequences of uncontrolled lipolysis. Despite reduced or absent adipose tissue, some lipodystrophic patients exhibit excessive lipid flux, severe insulin resistance, and fatty liver disease [5, 6]. Conversely, neutral lipid storage diseases are characterized by impaired lipolysis and lipid accumulation in tissues such as skeletal muscle yet typically lack the systemic features of metabolic syndrome [7]. Inflammatory and immune-mediated states can also disrupt lipolytic homeostasis; during sepsis, cancer, and critical illness, sustained cytokine signaling and sympathetic activation markedly increase lipolysis, promoting metabolic instability and tissue dysfunction [8,9,10].
A unifying feature across these conditions is dysregulated fatty acid flux rather than adiposity per se. Metabolic dysfunction arises in both obese and non-obese states, and in many cases impaired control of lipolysis contributes directly to insulin resistance independent of total fat mass [8,9,10,11]. Despite this, insulin resistance and diseases such as diabetes are frequently conflated with obesity, particularly in discussions of lipid metabolism. Lipolysis, for example, has been portrayed both as a causal driver of insulin resistance and as a desirable therapeutic target for weight loss [12,13,14]. This oversimplified framing reinforces the notion that increased fat mass is inherently pathogenic, overlooking a critical distinction: obesity often exists without uncontrolled lipolysis, and dysregulated lipolysis can occur in the absence of obesity.
Separating body weight from lipid homeostasis is therefore essential for understanding metabolic disease and identifying effective therapeutic strategies. This review aims to reframe lipolytic homeostasis as a key mechanism distinguishing obesity from metabolic dysfunction, with a particular emphasis on insulin sensitivity. We deliberately focus on the regulation of lipolysis and the disorders in which it is perturbed, rather than on adiposity itself. Our goal is to provide a conceptual framework in which lipolysis is understood as a process that can exacerbate metabolic disease when dysregulated, but when tightly controlled, can also be leveraged to improve metabolic health independent of body fat mass.
Regulators of the Enzymatic CascadeLipolytic flux is determined less by enzyme abundance than by tightly coordinated post-translational control, spatial organization, tissue specificity, and opposing hormonal signals. The canonical lipolytic pathway consists of three enzymes: adipose triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and monoacylglycerol lipase (MGL). Despite being identified most recently, ATGL catalyzes the initial and quantitatively dominant step, hydrolyzing triacylglycerols to diacylglycerols and accounting for most triglyceride hydrolysis in adipose tissue [15, 16]. Although ATGL mRNA expression is upregulated by fasting, glucocorticoids, Peroxisome Proliferator-Activated Receptor (PPAR) agonists, and Sirtuin 1–Forkhead Box 01 (SIRT1–FoxO1) signaling, and suppressed by insulin, feeding, and mechanistic Target of Rapamycin Complex 1 (mTORC1) activity, transcript levels do not reliably predict lipolytic activity [17]. β-Adrenergic stimulation and inflammatory cues such as TNF-α can suppress ATGL mRNA while increasing lipolysis, underscoring the primacy of post-translational regulation [18].
ATGL activity is enhanced by phosphorylation at serine 406 via AMP-activated protein kinase (AMPK) and by its obligate coactivator comparative gene identification 58 (CGI-58) [19, 20]. In the basal state, perilipin-1 coats lipid droplets and sequesters CGI-58, preventing ATGL activation. β-Adrenergic signaling activates protein kinase A (PKA), which phosphorylates perilipin-1 at multiple residues, including serine 517, releasing CGI-58 and enabling ATGL activation [21]. Disruption of this spatial control, as seen with pathogenic perilipin-1 mutations, causes unrestrained lipolysis, partial lipodystrophy, hypertriglyceridemia, and insulin resistance [22]. Additional layers of regulation emphasize tissue specificity: the ATGL inhibitor G0S2 is suppressed by fasting in adipose tissue but induced in liver [23, 24]. Other liver and adipose tissue differences include, how ATGL and HSL account for over 90% of lipolytic activity in white adipose tissue, whereas in fasted liver ATGL contributes less than half of triglyceride hydrolase activity; accordingly, ATGL deficiency causes hepatic steatosis without impairing VLDL secretion, indicating compensatory lipase activity [16, 25].
HSL catalyzes the rate-limiting step in diacylglycerol hydrolysis and is likewise regulated predominantly by post-translational and spatial mechanisms. HSL is activated by β-adrenergic signaling and inhibited by insulin, with phosphorylation by PKA at serine residues 660 and 663 serving as key activating events. Additional modulation occurs through AMPK, Extracellular signal-regulated kinase 1/2 (ERK), Ca2⁺/calmodulin-dependent kinase, and glycogen synthase kinase pathways [26, 27]. Full activation requires translocation of HSL to the lipid droplet, mediated by interaction with phosphorylated perilipin-1; coregulators such as Receptor interacting protein of 140 kDa (RIP-140) facilitate this process by promoting perilipin-1–HSL interactions [28]. Monoacylglycerol lipase (MGL) completes the cascade by hydrolyzing monoacylglycerols. MGL is ubiquitously expressed, localizes to membranes, cytoplasm, and lipid droplets, and is most abundant in adipose tissue. Genetic disruption of MGL impairs lipolysis and results in monoacylglycerol accumulation across tissues, confirming its essential role in lipid turnover [29].
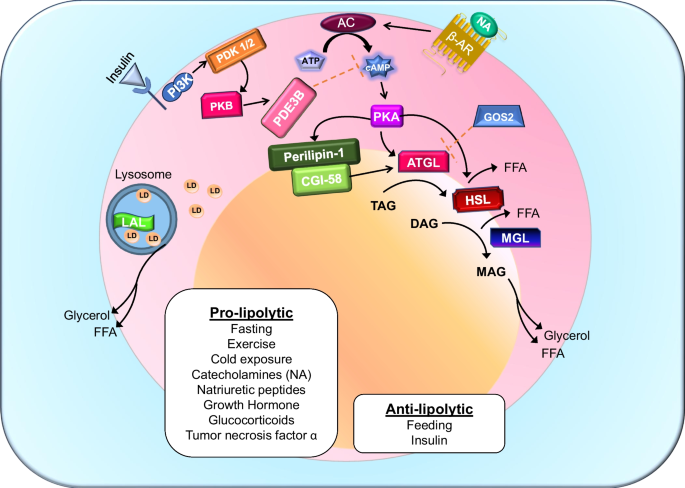
At the systemic level, insulin is the dominant antilipolytic hormone. At low physiological concentrations, insulin suppresses lipolysis through activation of phosphodiesterase 3B (PDE3B), reducing cAMP levels and attenuating PKA signaling, thereby preserving lipid droplet integrity [30]. Notably, insulin’s antilipolytic effects are more sensitive than its actions on glucose uptake, highlighting the priority of lipid containment in the fed state [31]. Insulin signaling within the central nervous system further restrains lipolysis by reducing sympathetic outflow to adipose tissue [32]. In contrast, catecholamines and glucagon stimulate lipolysis via cAMP–PKA signaling during fasting, cold exposure, exercise, and acute stress. These opposing inputs generate a pulsatile pattern of lipolysis that allows rapid, reversible mobilization of energy while minimizing lipid spillover and preserving metabolic flexibility. The core enzymes and regulatory nodes governing this cascade are summarized in Fig. 1.
Fig. 1 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Canonical and lysosomal regulation of adipocyte lipolysis. Pro-lipolytic stimuli including fasting, exercise, cold exposure, catecholamines (noradrenaline, NA), glucocorticoids, growth hormone, and tumor necrosis factor-α activate β-adrenergic receptors (β-AR), stimulating adenylyl cyclase (AC) to increase cAMP production and activate protein kinase A (PKA). PKA phosphorylates lipid droplet-associated proteins such as perilipin-1, promoting release of CGI-58 and activation of adipose triglyceride lipase (ATGL). Sequential hydrolysis of triacylglycerol (TAG) to diacylglycerol (DAG), monoacylglycerol (MAG), and ultimately glycerol and free fatty acids (FFAs) is mediated by ATGL, hormone-sensitive lipase (HSL), and monoacylglycerol lipase (MGL), respectively. Insulin and feeding exert anti-lipolytic control primarily through activation of phosphodiesterase 3B (PDE3B), reducing cAMP levels and attenuating PKA signaling, thereby preserving lipid droplet integrity and limiting fatty acid release. Lysosomal acid lipase (LAL) contributes to lipid mobilization via lipophagy, providing an additional layer of regulation independent of the canonical cytosolic lipase cascade. Figure adapted from review [33]. Abbreviations: β-AR, β-adrenergic receptor; NA, noradrenaline; AC, adenylyl cyclase; PKA, protein kinase A; PDE3B, phosphodiesterase 3B; ATGL, adipocyte triglyceride lipase; HSL, hormone-sensitive lipase; MGL, monoacylglycerol lipase; LAL, lysosomal acid lipase; TAG, triacylglycerol; DAG, diacylglycerol; MAG, monoacylglycerol; FFA, free fatty acid; PI3K, Phosphoinositide 3-kinase, GOS2, G₀/G₁ Switch Gene 2; PDK1/2, 3-phosphoinositide-dependent protein kinase; PKB, Protein Kinase B
Beyond the Canonical CascadeWhile the ATGL–HSL–MGL cascade defines canonical lipolysis, lipolytic flux and lipid remodeling are shaped by additional enzymatic pathways that operate across subcellular compartments and tissues. Lipid droplet–associated enzymes, redundant triglyceride hydrolases in non-adipose tissues, and lysosomal or autophagic lipolytic pathways together establish basal lipolytic tone and expand the regulatory capacity of lipid mobilization.
Several enzymes act directly at the lipid droplet alongside ATGL and HSL. PNPLA3, a member of the same protein family as ATGL, modulates lipid droplet remodeling and functions primarily in the fed state [34]. The common I148M variant (rs738409) exhibits reduced triglyceride hydrolase activity and increased retention at the lipid droplet surface, impairing lipid turnover and potentially restricting ATGL access to stored triglycerides [35]. This defect promotes hepatic triglyceride accumulation and increases susceptibility to steatosis and fibrosis, often independent of overall adiposity or elevated circulating fatty acids, highlighting how local lipid handling can drive tissue-specific pathology [35].
Other noncanonical lipases contribute to lipid signaling and metabolism outside adipose tissue. ABHD6 (α/β-hydrolase domain–containing protein 6) is a serine hydrolase localized primarily to the endoplasmic reticulum and, to a lesser extent, lipid droplets. Although it partially overlaps with MGL in hydrolyzing monoacylglycerols, ABHD6 plays a more prominent role in non-adipose tissues, particularly the liver and central nervous system [36]. By regulating monoacylglycerol pools—including the endocannabinoid 2-arachidonoylglycerol (2-AG)—ABHD6 links lipid metabolism to signaling pathways [37]. In metabolic tissues ABHD6 inhibition improves insulin sensitivity and hepatic lipid accumulation in diet-induced obesity models, underscoring its role in coupling lipid turnover to metabolic regulation [38].
Lipolytic dominance also varies markedly by tissue. In the liver, triglyceride hydrolysis relies on enzymes beyond the canonical cascade, including carboxylesterases such as CES1. CES1 contributes to hepatic triglyceride hydrolysis and can partially compensate for ATGL deficiency [39, 40]. Unlike ATGL, which operates at the cytosolic surface of lipid droplets and is tightly regulated by β-adrenergic and insulin signaling, CES1 functions within the endoplasmic reticulum. There, it participates in the mobilization of triglycerides destined for very-low-density lipoprotein (VLDL) assembly and secretion, positioning CES1 at the interface between intracellular lipid mobilization and lipoprotein production [41].
Not all lipid mobilization occurs at neutral pH. LIPA encodes lysosomal acid lipase (LAL), which hydrolyzes cholesteryl esters and triacylglycerols within lysosomes as part of the lipophagic pathway. Unlike ATGL and HSL, which act on cytosolic lipid droplets, LAL functions downstream of autophagy following delivery of lipid droplets to lysosomes [42]. Adipocyte LIPA expression and activity increase during prolonged fasting, cold exposure, and β-adrenergic stimulation; inhibition of LAL blunts fasting-induced rises in plasma free fatty acids and impairs thermogenesis and oxygen consumption, demonstrating its importance for lipid mobilization [43]. In hepatocytes, LAL-driven lipophagic flux supplies fatty acids for oxidation and integrates lipid turnover with cholesterol sensing and trafficking pathways [44]. Because LAL activity is regulated by autophagic flux and lysosomal biogenesis programs, including MiT/TFE family transcription factors, this pathway is highly sensitive to nutrient state and energy stress [45]. Loss of LIPA causes Wolman disease or cholesteryl ester storage disease, characterized by severe hepatic lipid accumulation and systemic metabolic dysfunction [46]. Lysosomal lipolysis remains less characterized, and its relative contribution—whether complementary to the canonical pathway or essential during high energy demand—has yet to be defined.
Obesity and LipolysisObesity is metabolically heterogeneous. As summarized in Table 1, metabolic outcomes across diverse disease states are more accurately organized along a spectrum defined by lipolytic regulation rather than adiposity alone. Both low- and high-fat states can exhibit pathology when lipolytic control is lost. Although increased fat mass defines obesity, only a subset of individuals with obesity display dysregulated lipolysis [4]. Individuals who retain normal metabolic parameters despite obesity are classified as metabolically healthy obese (MHO), a phenotype observed in approximately 10–30% of obese populations and therefore not rare. Diagnostic criteria for MHO include a BMI ≥ 30 kg/m2, fasting triglycerides ≤ 1.7 mmol/L, HDL cholesterol > 1.0 mmol/L in men or > 1.3 mmol/L in women, and blood pressure ≤ 130/85 mmHg [4].
Table 1 Human diseases and physiological states organized by direction of lipolytic activity and adiposityFor many individuals, however, metabolic health in obesity is transient. One of the earliest abnormalities preceding overt metabolic disease is impaired insulin-mediated suppression of lipolysis, which frequently emerges before detectable hyperglycemia [54]. In this state, basal lipolytic tone remains inappropriately elevated despite circulating insulin, resulting in chronic low-level fatty acid release [55]. Concurrently, obesity-associated inflammation promotes catecholamine resistance through downregulation of β-adrenergic signaling components, impairing stimulated lipolysis [56]. Thus, adipocytes in metabolic dysfunction often display a paradoxical phenotype characterized by inadequate suppression of basal lipolysis alongside impaired execution of stimulated lipolysis. These defects reflect a loss of regulatory precision rather than a simple excess of fat mass.
Excess fatty acid flux contributes to insulin resistance in part through the accumulation of bioactive lipid intermediates. Elevated free fatty acids (FFAs) impair insulin signaling, disrupt mitochondrial function, and activate inflammatory pathways [57]. In liver and skeletal muscle, accumulation of diacylglycerols and ceramides interferes with insulin signaling cascades [58]. In pancreatic β-cells, chronic exposure to fatty acids—particularly palmitate and ceramides—induces endoplasmic reticulum stress, oxidative injury, inflammation, and secretory dysfunction, accelerating β-cell failure [59]. Insulin resistance correlates strongly with circulating non-esterified fatty acids and bioactive lipid intermediates [60], reinforcing a self-sustaining cycle of dysregulated lipolysis and metabolic decline.
The anatomical source of lipolysis further modulates metabolic risk. Visceral adipose tissue is particularly pathogenic due to higher basal lipolytic activity, reduced insulin sensitivity, increased inflammatory signaling, and direct portal drainage to the liver. This portal delivery of fatty acids promotes hepatic gluconeogenesis, triglyceride synthesis, and very-low-density lipoprotein (VLDL) secretion, amplifying metabolic dysfunction more acutely than subcutaneous adipose tissue [61]. Experimental activation of adipocyte lipolysis acutely increases circulating FFAs to approximately 1.5 mM within 30 min, followed by a comparable rise in circulating triglycerides. In contrast, hepatic triglyceride levels peak later, approximately three hours after stimulation, and remain elevated for over 12 h despite normalization of circulating lipids. When adipocyte fatty acid release is impaired, as in mice lacking ATGL specifically in adipocytes, this hepatic triglyceride accumulation does not occur [62]. These findings are particularly relevant given the rising prevalence of metabolic function-associated fatty liver disease (MAFLD).
Importantly, triglycerides themselves are metabolically inert storage molecules rather than signaling toxins. Esterification of fatty acids into triglycerides neutralizes their detergent-like properties and limits conversion into bioactive lipid intermediates. Lipid storage therefore functions as a protective buffering system that accommodates fluctuations in nutrient availability. This buffering capacity is mediated by lipid droplets across multiple tissues, including adipose tissue, liver, and skeletal muscle [2, 3]. Lipid droplets are dynamic organelles equipped with regulatory proteins and enzymatic machinery that govern lipid sequestration and mobilization. Effective lipid containment within adipose tissue shields liver, muscle, and pancreas from lipid overload; it is the failure of this containment system—rather than triglyceride accumulation per se—that poses the primary metabolic threat associated with insulin resistance [2, 3].
Multiple lines of evidence challenge a causal relationship between adiposity and insulin resistance. The “athlete’s paradox” demonstrates that endurance-trained individuals exhibit elevated intramyocellular triglyceride content while remaining highly insulin sensitive. Subsequent studies show that exercise in older obese adults improves insulin sensitivity alongside favorable lipid partitioning, independent of weight loss [63]. The primacy of lipid handling over adiposity is further supported by transplantation studies in which subcutaneous adipose tissue from exercised mice confers marked improvements in insulin sensitivity when transplanted into sedentary, high-fat diet–fed obese mice [64]. Similarly, ATGL-, HSL-, and MGL-deficient mice accumulate large triglyceride stores equivalent to wild-type mice on a high-fat diet, yet maintain preserved or even improved insulin sensitivity, likely due to reduced systemic fatty acid exposure [29, 65, 66]. In humans, neutral lipid storage diseases—including neutral lipid storage disease with myopathy and with ichthyosis—feature extensive triglyceride accumulation across tissues without a corresponding requirement for diabetes or severe insulin resistance [7]. Collectively, these examples demonstrate that triglyceride burden alone is insufficient to drive insulin resistance.
Conversely, diminished metabolic health does not require excessive adiposity. Compared with European populations, Asian cohorts with type 2 diabetes develop disease at lower mean body mass index and are characterized by early β-cell dysfunction [11]. These observations further support the concept that insulin resistance is fundamentally a disorder of lipid flux regulation rather than adiposity.
Disorders marked by reduced adipose tissue mass frequently exhibit profound metabolic pathology. Congenital generalized and partial lipodystrophies, characterized by severely limited adipose storage capacity, are associated with uncontrolled fatty acid flux, ectopic lipid deposition, severe insulin resistance, and fatty liver disease [5, 6, 67]. Similarly, pathogenic mutations in perilipin-1 (PLIN1), which disrupt lipid droplet regulation and CGI-58 sequestration, lead to unrestrained lipolysis, partial lipodystrophy, and metabolic syndrome despite reduced or redistributed fat mass [22].
Hyperlipolytic states further dissociate adiposity from metabolic outcomes. In cancer cachexia and experimental sepsis, adipose tissue mass declines while lipolysis remains chronically elevated due to inflammatory cytokine signaling and heightened sympathetic activation. Sustained fatty acid release under these conditions contributes to systemic insulin resistance, hepatic lipid overload, and multi-organ dysfunction, despite low or declining adiposity [
Comments (0)