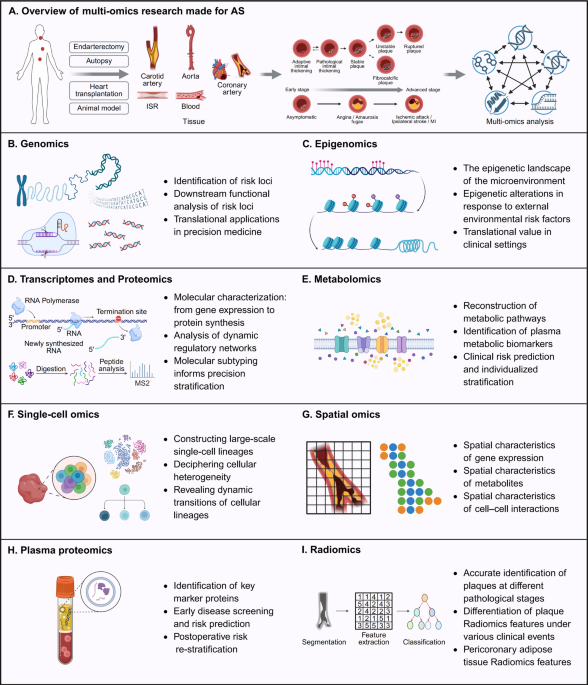
Single-cell Transcriptomics and Epigenomics
Among emerging technologies, scRNA-seq allows the high-resolution profiling of thousands of individual cells, revealing their gene-expression programs in an unbiased and systematic manner. In atherosclerosis, scRNA-seq has uncovered previously unknown subtypes of macrophages, SMCs, endothelial cells (ECs), and T cells, each with potentially distinct roles in inflammation, lipid metabolism, and matrix remodeling. These findings challenge conventional categorizations and point to a previously unsuspected degree of cellular heterogeneity and dynamic cellular interactions [6].
Additionally, scATAC-seq generates important regulatory maps. This technique provides knowledge on chromatin accessibility, offering insight into regulatory elements, enhancer-promoter interactions, and the activity of transcription factors. When applied to atherosclerotic tissues, scATAC-seq has confirmed key epigenetic regulators of lineage reprogramming, such as KLF4 and TCF21, which control SMC plasticity and macrophage activation [7]. Such gene regulatory networks are critical patterns for understanding gene regulation and the intricate interactions that drive biological processes. Recent advances have highlighted the potential of integrating scATAC-seq to scRNA-seq in offering unprecedented insights into how chromatin accessibility plays an important part in gene regulation and its role in pathological transitions [8].
Multiplexed Protein and Multimodal Profiling
Multiplexed imaging plataforms such as Imaging Mass Cytometry (IMC), CODEX, and Phenocycler allow simultaneous detection of dozens of protein markers. These tools enhance immune phenotyping and validate transcriptional data. Emerging single-cell multi-omics technologies have evolved to integrate multiple molecular modalities simultaneously, encompassing the transcriptome, genome, epigenome, epitranscriptome, proteome, and metabolome. These techniques bridge the gap between transcriptomic and proteomic data enhancing cell state resolution and facilitate better functional annotation of immune and vascular cell populations [9, 10].
Recent progresses in technologies, like REAP-seq and ECCITE-seq, allow simultaneous measurement of proteins and mRNAs in single cells. Hopefully, molecular data on clonotype information (e.g., T-cell receptor (TCR) / B-cell receptor (BCR) sequences, perturbations (e.g., CRISPR-based screens), or protein secretion are obtained [11]. These capabilities expand the scope of single-cell analysis from observational to mechanistic studies, potentially accelerating drug discovery and biomarker elaboration. The diversity of TCRs and BCRs underpins the adaptive immune system’s ability to recognize and respond to a wide array of antigens. Recent advancements in RNA sequencing have expanded its application beyond transcriptomics to include the analysis of immune repertoires, enabling the exploration of TCR and BCR sequences across various physiological and pathological contexts, such as atherosclerosis [12].
Spatial Omics
A critical limitation of dissociative single-cell techniques is the loss of spatial context, which obscures the spatial relationships between cells and their microenvironment. Spatial transcriptomics overcomes this drawback by preserving tissue architecture while allowing high-resolution gene expression outlining. This methodology provides a more integrated view of cellular heterogeneity, intercellular interactions, and tissue architecture [13].
Spatial transcriptomics technologies can be broadly categorized into sequencing based and probe-based approaches. While sequencing based platforms such as Visium (10 × Genomics), Stereo seq (BGI), and Aviti (Element Biosciences) enable transcriptome wide profiling with high sensitivity, they often lack single cell or subcellular resolution, limiting their ability to precisely map cellular interactions within complex tissues.
In contrast, probe-based platforms such as Xenium (10 × Genomics), CosMx SMI (NanoString), and MERSCOPE (Vizgen) offer subcellular resolution and enable highly multiplexed detection of targeted transcripts, making them particularly well suited for dissecting the spatial organization of cell states and interactions within the tissue microenvironment.
When applied to atherosclerotic plaques and perivascular regions, these high-resolution imaging tools allow detailed characterization of cellular heterogeneity – such as macrophage-rich necrotic cores or endothelial clusters undergoing endothelial-to-mesenchymal transition (EndMT) – and how they are spatially distributed and engage in paracrine signaling, identification of rare or novel cell states, and spatial mapping of receptor–ligand interactions. Such analyses can uncover key mechanistic pathways involved in disease progression and immune modulation in atherosclerosis [14, 15].
Spatial proteomics is a multidimensional technique that studies the spatial distribution and function of proteins within cells or tissues across both spatial and temporal dimensions. This approach complements transcriptomic data and allows for the direct visualization of protein expression within intact tissue sections, providing insights into the cellular and molecular organization of atherosclerotic plaques. Compared to transcriptomics, protein markers are more stable and better defined for immune cell phenotyping, which is particularly important in the inflamed and heterogeneous environment of atherosclerosis. This consideration makes spatial proteomics a more robust approach for identifying and classifying immune and stromal populations involved in plaque progression and remodeling.
In addition to functional profiling, spatial proteomics plays a critical role in cell segmentation. The use of membrane and nuclear markers enables accurate definition of single-cell boundaries, which is essential for generating high-quality single-cell spatial datasets. Proper segmentation ensures that molecular signals are assigned to the correct cell, which is crucial for downstream analyses such as cell–cell interaction mapping and neighborhood analysis.
While spatial transcriptomics provides broader coverage of gene expression, spatial proteomics offers higher spatial resolution and improved accuracy in defining cell types and states. In the context of atherosclerosis, where immune infiltration, endothelial dysfunction, and smooth muscle plasticity occur in highly organized microenvironments, spatial proteomics provides a more precise and functionally relevant view of the cellular landscape driving disease. Combining scRNAseq data and spatial transcriptomics enable the generation of high-resolution maps of human atherosclerotic plaques [1].
Future Directions in Spatial Analyses
Single-slide spatial multiomics enables the simultaneous analysis of RNA and protein expression within the same tissue section, preserving spatial context while integrating molecular modalities. This approach enhances cell type annotation by leveraging the stability and specificity of protein markers for accurate segmentation, particularly in immune and vascular cells. Once cells are segmented and classified based on proteomic data, transcriptomic profiles can be used to reveal transcriptional heterogeneity and functional states within each population. In the context of atherosclerosis, this strategy is powerful, allowing the identification of distinct immune and stromal subsets and enabling the reconstruction of cell–cell communication networks. The ability to assign molecular identity and interaction potential within the same spatial framework makes single slide multiomics a pivotal tool for elucidating the cellular architecture and signaling landscapes of complex tissue microenvironments.



Comments (0)