
Remember me
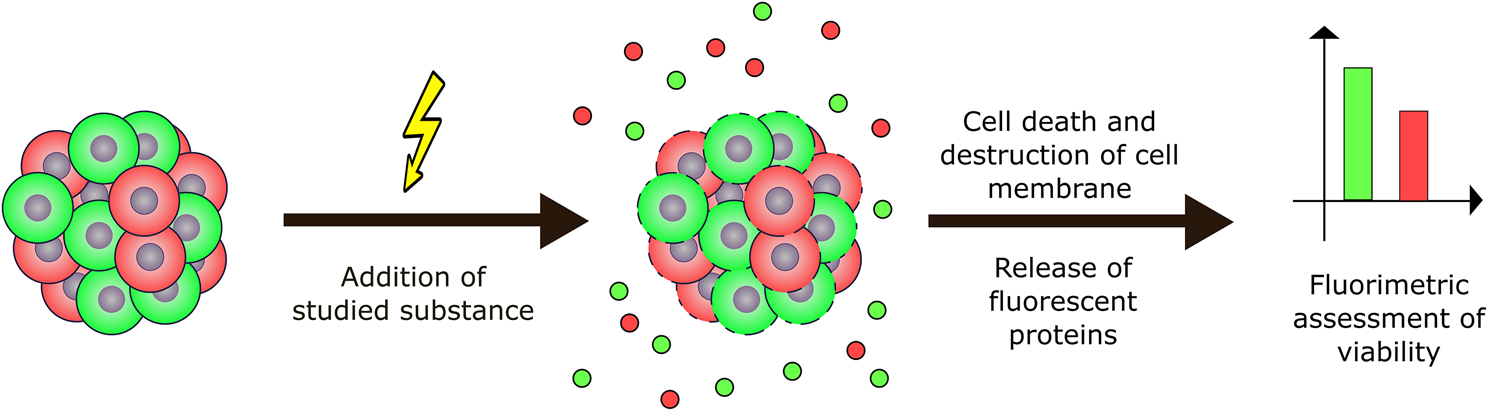


For this experiment, cell lines NCI-H1299-RFP and Huh7-GFP were taken. The spheroids were obtained by the hanging drop method. Briefly, rows of drops with a volume of 25 μl were placed on the inside of the top lid of a Petri dish, each drop containing 2000 cells. 10 ml of PBS was then added to the bottom of the Petri dish and the top lid was carefully inverted and placed on top. After 5 days, the spheroids were formed.
The medium was then changed twice, 10 μL at a time. Then after, 5th day, 10 μl of medium in droplets, which contained spheroids for the experiment, was replaced by 10 μl of complete culture medium containing 375 μM capecitabine. Thus, the final concentration of capecitabine in 25 μl was 150 μM. This concentration was chosen based on the results of a previous experiment.
After 48 h of incubation in a humidified CO2 incubator at 37 °C and 5% CO2, 10 μl of medium was taken from each group of droplets and pooled into their respective wells. The entire contents of the drops designated as “negative viability control” were taken along with the spheroids and transferred into a 250 μl PCR tube. 6 µl of Triton-X100 was added there. The tube was then vortexed and left to incubate on a rotator at room temperature for half an hour before being mixed again and centrifuged at 1000g for 10 min to pellet debris. 60 µl from this tube was transferred to the corresponding well of the plate. This new plate was then measured on the BMG Clariostar plate reader in fluorimetry mode. The results of the experiment are shown in Fig. 6a–c.
Alginate hydrogelFor the purposes of this work, an experiment was conducted to create three-dimensional “droplets” of cross-linked sodium alginate with cells enclosed inside. The protocol outlined in the work [27] with minor modifications was taken as a basis. The volume of spheroids was changed—100 μl, the order of addition and composition of the cross-linking solution CaCl2, the concentration of sodium alginate and the number of cells were changed. The sodium alginate concentration of 2.5% stated in Smit et al. [27] turned out to be irreproducible in practice—uniform stirring and manipulation of such a solution turned out to be impossible due to its excessive viscosity. After a series of preliminary experiments, it was decided to settle on a concentration of 1.1%, and use 1% CaCl2 in F12 as a crosslinking solution.
Before starting the experiment, a solution of 1.1% sodium alginate in Hanks’ buffer was prepared. 1.1 grams of sodium alginate was added to 100 ml of Hanks’ solution in a heat-resistant flask. Stirring was carried out with a magnetic armature for 4 h at a temperature of 55 °C. After stirring all visible lumps and inhomogeneities, the temperature on the magnetic stirrer was raised to 200 °C and the solution was brought to a boil twice for sterilization (since its sterilization through filters is impossible due to its viscosity). After which the flask with the solution was introduced into the laminar flow and the solution was aseptically transferred into a test tube for cooling.
A solution for “crosslinking” was also prepared—F12 with the addition of sterile calcium chloride to a concentration of 1%. To perform this experiment, cell lines NCI-H1299-RFP, NCI-H1299-GFP, Huh7-GFP and Huh7 were taken. Cell cultivation was carried out according to standard procedures. Upon reaching 80–90% confluency, Petri dishes with cultures were washed with versene, the cells were trypsinized with a 0.25% trypsin-EDTA solution, and the cell suspension was transferred to centrifuge tubes. After centrifugation at 1000 g for 5 min, the supernatant was removed and the cells were resuspended in 2 ml of fresh F12 medium. The concentration of the suspensions in all cell lines was adjusted to 1 million/ml, and 1 ml of the suspension of each line was transferred into a 1.5 ml Eppendorf, in the case of cocultures—0.5 ml of each.
Thus, the following were prepared for the experiment:
1)Monocultures—NCI-H1299-GFP, NCI-H1299-RFP, Huh7-GFP
2)Cocultures—NCI-H1299-GFP/Huh7, NCI-H1299-RFP/Huh7 and NCI-H1299/Huh7-GFP.
After pippeting, tubes with cell suspensions were centrifuged at 1000g for 10 min. In parallel, two 12-well plates were taken, and 4 ml of cross-linking solution was added to each well. After centrifugation, the supernatant was removed and 1 ml of Hanks’ solution with sodium alginate was added to each tube. After slow and careful mixing, 2 drops of a cell suspension in sodium alginate were dripped into the wells of the plate from a height of 5–7 cm. The volume of each drop is 100 µl. This height is optimal for such viscous droplets that they acquire a spherical shape upon contact with the cross-linking solution. The excavation was carried out according to the following scheme indicated in the Table 1.
Table 1 Scheme of sodium alginate dropletsThe crosslinking solution was then removed from all wells, the wells were washed twice with F12, and 2 ml of fresh F12 was added to each well. 3D cultures in alginate drops were cultivated for 14 days, the medium was replaced every 2 days. As visible three-dimensional structures grew and formed in the alginate droplets (shown in the figures), the formation of spheroids was monitored Fig. 2).
Fig. 2
a NCI-H1299-GFP culture growth, phase contrast microscopy. 5th day of cultivation, b NCI-H1299-GFP culture growth, GFP fluorescence channel. 400x magnification
Interestingly, the Huh7 and Huh7-GFP cultures, when growing in an alginate matrix, along with spheroids (at the later stages of cultivation—days 9–12) began to form elongated, radially located formations (Fig. 3).
Fig. 3
Huh7 culture, 11th day of cultivation, phase contrast microscopy. 100x magnification
On the 14th day of cultivation, the medium in the wells was replaced with a medium containing 150 μM capecitabine (for the corresponding wells). In control wells, the medium was replaced with regular F12 medium. After 36 h, 20 μl of Triton X100 was added to the negative control wells. After another 12 h, 100 μl of medium was taken from each well of each culture into the wells of a 96-well plate, and fluorescence was measured in the GFP and RFP channels on a CLARIOStar BMG spectrofluorimeter. Another day later, an additional measurement was performed. Thus, we got 2 time points—48 and 72 h. The results of the experiment are shown in Fig. 7a–f.
Demonstration of the limits of applicability of the method and its use for assessing the cytotoxicity of other compoundsTo identify the limits of the method’s applicability, we conducted another experiments—
The first one was to establish how heavy metals (Co, Pt) affect the measured fluorescence intensity. As is known from publications [28], platinum compounds can inhibit LDH and bias the results of viability analyses which based on measuring its activity. In addition, cobalt can also inhibit this enzyme. However, in the method described in this article, the reporter proteins are fluorescent proteins, and we conducted the experiment with them.
In a second experiment, we measured how drug metabolites themselves can bias viability measurements. For this experiment, we used paracetamol as a model compound. It was chosen because its main metabolite, NAPQI (N-acetyl-p-benzoquinone imine), is capable of depleting glutathione stores and forming conjugates with proteins, thus inhibiting them and/or disrupting their spatial structure.
Experiment to determine the influence of heavy metals on measurement resultsNCI-H1299-GFP and NCI-H1299-RFP cells were prepared for the experiment and lysed using the same protocol as described in the chapter “Design of proof-of-concept experiment”. Then, 1 M CoCl2 solution was prepared in serum-free medium, a solution of carboplatin with a concentration of 10 mg/ml was also taken.
50 μl of lysate were added to the wells of 96-well plates, in groups—lysate of cells expressing GFP, and lysate of cells expressing RFP. Then 50 μl of solutions of the studied substances were added to these wells (final concentrations are indicated): CoCl2 from 500 to 0.5 mM, carboplatin—from 1000 to 10 μg/ml. Wells in which 50 μl of serum-free medium was added to the lysate were used as a positive control; wells without lysate, which contained 100 μl of serum-free medium, were used as a blank. The plates were mixed on an orbital shaker and then incubated at 37 °C overnight. After that, fluorescence was measured in the GFP and RFP channels. The results of the experiment are shown in Fig. 8 (for CoCl2) and 9 (for carboplatin).
Experiment to assess the effect of drug metabolites on fluorescent proteinsHuh7, Huh7-GFP, NCI-H1299-GFP and NCI-H1299-RFP cells were prepared for the experiment using the same protocol as described in the chapter “Experiment to determine the toxicity of capecitabine using described method”. The cells were seeded in a 96-well plate at 10,000 cells/well in the same combinations. After 24 h, paracetamol was added to the cells at final concentrations of 300 to 10 mM. After another 48 h, the culture medium was taken for fluorimetry in GFP and RFP channels. The results of the experiment are shown in Fig. 10a–c.
Comments (0)