
Remember me
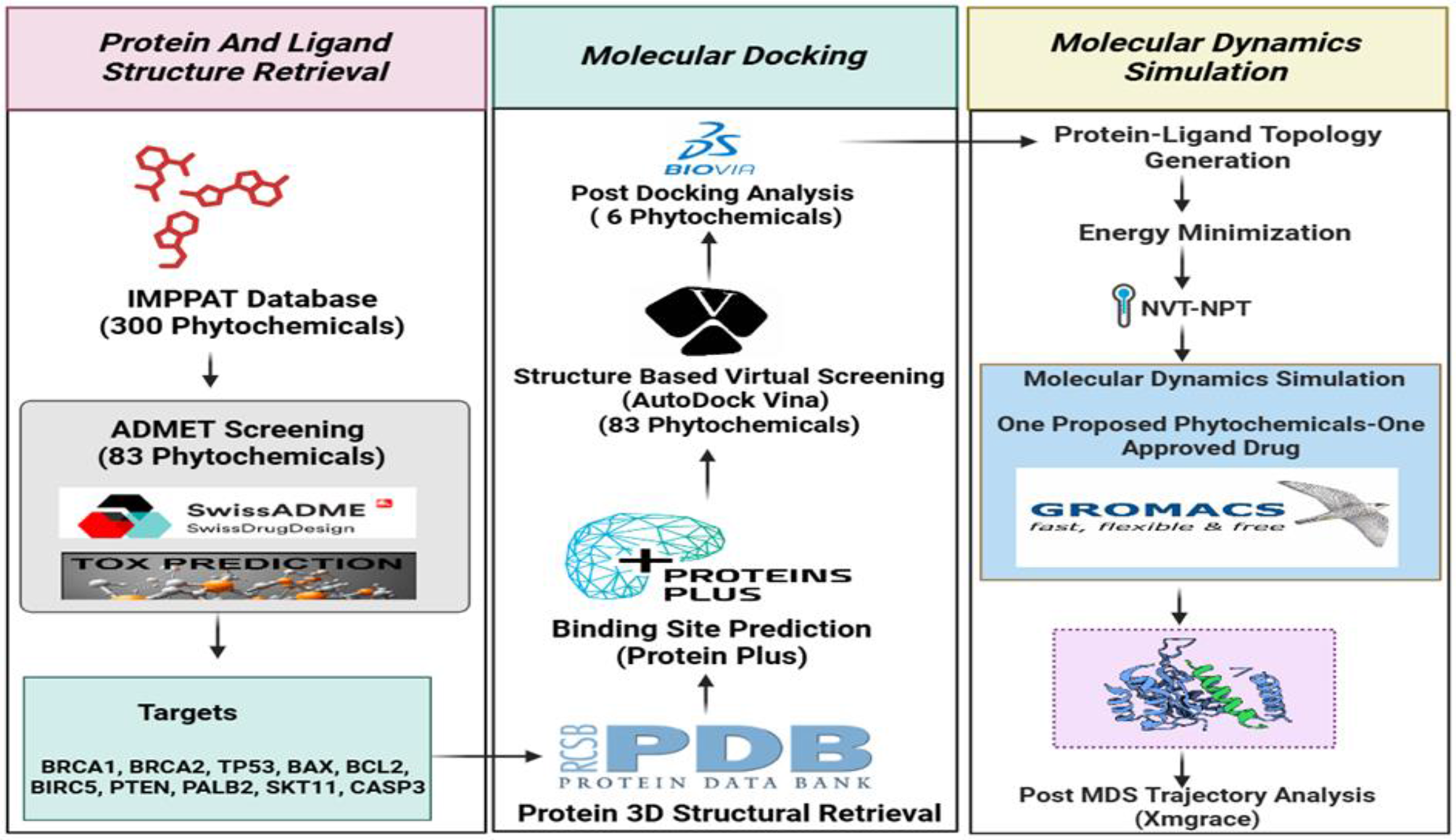
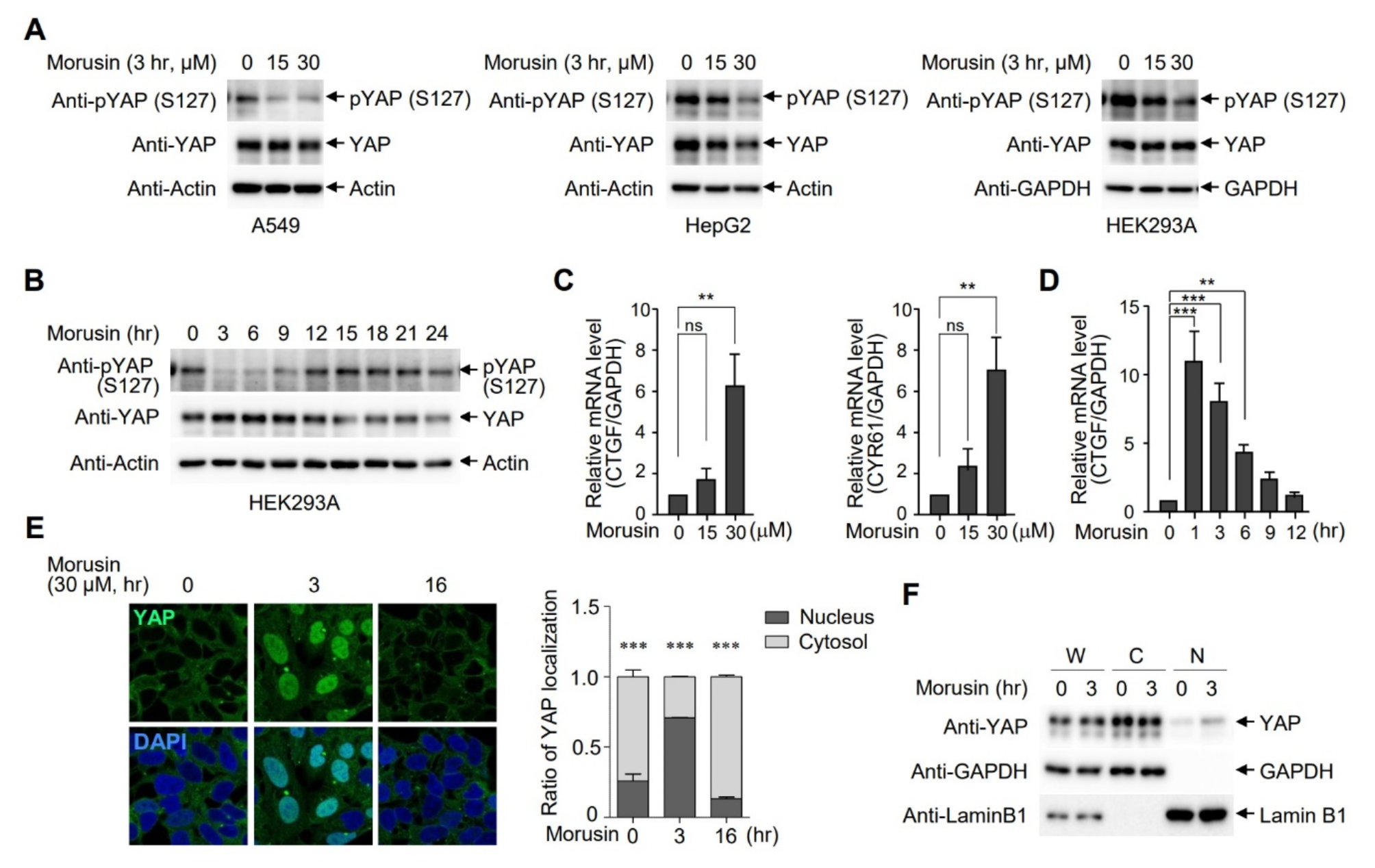


The ligand repository consisted of 300 phytochemicals, including flavonoids, lignans, phenolic acids, stilbenes, and non-phenolic metabolites retrieved from the IMPPAT 2.0 database. All these 300 phytochemicals were passed through SwissADME (Lipinski rule of five, gastrointestinal absorption, blood-brain barrier (BBB) permeability, P-glycoprotein (PGP) substrate interaction) and the Protox-II web tool (carcinogenicity, cytotoxicity, immunotoxicity, and mutagenicity). A total of 83 phytochemicals passed the ADMET parameters. These compounds were extracted from the PubChem database in three-dimensional SDF file format. They were then processed using Open Babel GUI to generate pdbqt input files for use in AutoDock Vina. A detailed methodology has been already discussed in the methodology section.
Protein retrievalAll protein molecules were meticulously chosen through an extensive review of the literature and subsequently acquired from the Protein Data Bank (PDB). The nomenclature and corresponding PDB identifiers for the selected proteins are as follows: BRCA1 (PDB ID: 1T15), BRCA2 (PDB ID: 3EU7), PALB2 (PDB ID: 2W18), PTEN (PDB ID: 1D5R), TP53 (PDB ID: 4MZI), STK11 (PDB ID: 5WXN), BCL2 (PDB ID: 6O0K), BAX (PDB ID: 2G5B), BIRC5 (PDB ID: 2QFA), and CAPS3 (PDB ID: 1NME), as illustrated in Fig. 2.
Fig. 2
3D Structures of the crystal structure of all protein structure molecules. The figure depicts the 3D structures of several proteins implicated in cancer development and progression. Each protein is labeled accordingly
Active site prediction and protein PreparationAll proteins examined in the present investigation were analyzed using the ProteinPlus web server, which facilitated the prediction of the binding sites for each respective residue. A comprehensive account of all proteins, including but not limited to PDB identification numbers, the resolution of the deposited structures, graphical representations, surface area, volume, and the number of residues constituting the ligand binding pocket, is provided in the supplementary material (Table S3).
Protein-ligand molecular DockingProtein preparation and grid generation procedures were conducted by established protocols. The co-crystallized ligand (Venetoclax) was re-docked in the experimentally proven binding pocket of BCL2 (PDB ID: 6O0K) protein to validate the docking protocol followed throughout the study. This analysis revealed the value of RMSD value of 0.72 Å upon superimposition of the co-crystallized ligand with a docked pose. The values were found within the acceptable range of RMSD i.e., < 2.0 Å. This step ensures the correct docking procedure for other protein targets as well (Fig. 3).
Fig. 3
Re-docking and superimposition of co-crystallized ligand (Pink) and docked pose (Blue) in the experimentally proven binding pocket of BCL2 (PDB ID: 6O0K) protein
The dimensions of the grids (Table S3) corresponding to the individual proteins were input into the AutoDock Vina software to evaluate a ligand library composed of 83 phytochemicals alongside the FDA-approved drug Olaparib. Upon completion of the docking simulations conducted in triplicate, an average docking score was computed for each of the proposed compounds as well as for the approved compound. Subsequently, these docking scores were subjected to analysis utilizing the BIOVIA Discovery Studio visualizer. Table 1 illustrates the average docking scores for all protein targets in conjunction with the proposed ligands, which demonstrate docking scores surpassing that of the approved ligand about the respective protein. The results indicate that the proposed compounds Ajmaline, Bayogenin, Rotenone, Tephrosin, and alpha-Toxicarol exhibited superior performance compared to the approved compound (Olaparib) about docking scores assessed by the AutoDock Vina software across various receptors.
Table 1 Docking score (kcal/mol) of hit compounds with all the protein targetsBinding energy is a critical parameter in molecular docking studies, as it quantifies the strength and stability of interactions between a ligand and a target protein. Lower binding energies indicate stronger and more stable interactions, which are often associated with higher binding affinity. In a therapeutic context, this suggests that the phytochemical has the potential to effectively engage with its target proteins, enhancing its efficacy as a drug candidate.
A comprehensive evaluation of the docking scores revealed that Bayogenin achieved scores of -9.4 kcal/mol, -7.9 kcal/mol, and − 9.3 kcal/mol, while the approved drug Olaparib registered scores of -9.4 kcal/mol, -7.6 kcal/mol, and − 7.7 kcal/mol against the three protein targets BRCA2, BAX, and PALB2, respectively. The proposed compounds Rotenone, Tephrosin, and alpha-toxicarol exhibited higher docking scores than the approved compounds across two protein targets. The docking score for Rotenone was recorded at -9.1 kcal/mol and − 8.2 kcal/mol, whereas Olaparib displayed scores of -9 kcal/mol and − 7.6 kcal/mol for the protein targets BCL2 and BAX, respectively. The docking score for Tephrosin was noted as -7.7 kcal/mol and − 8.9 kcal/mol, while Olaparib presented scores of -7.6 kcal/mol and − 7.7 kcal/mol for the protein targets BAX and PALB2, respectively. Likewise, alpha-Toxicarol yielded docking scores of -8 kcal/mol and − 8.6 kcal/mol, whereas Olaparib was measured at -7.6 kcal/mol and − 7.7 kcal/mol for the protein targets BAX and PALB2, respectively. Another noteworthy compound, Ajmaline, obtained a docking score of -7.8 kcal/mol, whereas Olaparib recorded a score of -7.7 kcal/mol for the protein target PALB2.
Furthermore, through the examination of docked poses utilizing the Discovery Studio analyzer, it was determined that Bayogenin exhibited a higher frequency of conventional hydrogen bonds in comparison to the approved compound across multiple sites. The BRCA2-Bayogenin complex demonstrated the formation of three conventional hydrogen bonds involving the amino acid residues Gln921, Val969, and Asp1125. Conversely, the BAX-Olaparib complex established merely one conventional hydrogen bond with the Phe876 amino acid residue, which facilitated Bayogenin’s superiority over Olaparib. The BAX-Bayogenin complex manifested two conventional hydrogen bonds with the residues Gln39 and Tyr91, while the BAX-Olaparib complex presented two conventional hydrogen bonds at the residues Ser43 and Trp101. Furthermore, additional interaction modalities were also identified, which rendered the BAX-Olaparib complex more favorable at this juncture compared to the BAX-Bayogenin complex. The PALB2-Bayogenin complex displayed three conventional hydrogen bonds at Glu1018, His1061, and Lys1062, in contrast to the PALB2-Olaparib complex, which exhibited two conventional hydrogen bonds at the residues Gln921 and Glu1018, thereby again establishing the superiority of the PALB2-Bayogenin complex over the PALB2-Olaparib complex. Other significant interactions within the respective protein-ligand complexes have been delineated in (Table 2). A detailed description of the ADMET predictions of the filtered compounded has been shown in Table S1 and Table S2. Furthermore, representations of both 2D interactions among the respective protein-ligand complexes are illustrated in Fig. 4.
Table 2 Molecular interactions of BRCA-2, BAX, and PALB-2 with Bayogenin and OlaparibFig. 4
Schematic 2D representation of binding residue configuration in the docked protein-ligand complexes (a) BRCA2-Bayogenin, (b)BRCA2-Olaparib, (c)BAX-Bayogenin, (d) BAX-Olaparib, (e) PALB2-Bayogenin and (f) PALB2-Olaparib
Upon evaluating the hit compounds, it was discerned that Bayogenin demonstrated relatively superior performance compared to the other hit compounds, as evidenced by its higher or nearly equivalent docking scores about the approved drug. Bayogenin excelled across three targets, while the other hits were effective on only two or one targets, as elaborated previously. The quantity of conventional hydrogen bonds further substantiated Bayogenin advantage over the approved compound. Given the intense competition between the shortlisted drug (Bayogenin) and the approved drug (Olaparib), there arose a necessity for additional analysis, which was subsequently conducted through MDS.
Molecular dynamics simulation (MDS) studiesMolecular Dynamics Simulation (MDS) simulations provide insights into the stability of these protein-ligand complexes under physiological conditions. A stable interaction over time suggests that the compound can maintain its binding to the target protein despite environmental fluctuations. Proteins within a solution milieu exhibit stochastic migration across a designated temporal spectrum while displaying pronounced oscillatory behaviors. MDS provides a more reliable assessment of the stability of protein-ligand complexes compared to traditional molecular docking approaches. A time-resolved MDS was performed for a duration of 200 ns concerning six protein-ligand complexes delineated during the preliminary virtual screening phases, specifically BRCA2-Bayogenin, BRCA2-Olaparib, BAX-Bayogenin, BAX-Olaparib, PALB2-Bayogenin, and PALB2-Olaparib, utilizing Gromacs-23.1 software.
RMSD trajectory analysisThe Root Mean Square Deviation (RMSD) plot provides a quantitative assessment of the extent of structural deviation exhibited by an atomic group relative to its original reference structure, as it evolves to its final conformation throughout the simulation. Generally, diminished RMSD values are indicative of enhanced stability, while elevated values suggest a decrease in stability. To scrutinize the stability of six protein-ligand complexes and elucidate the behavior of these systems, the RMSD values were meticulously evaluated over time. The accompanying Fig. 5. illustrates the RMSD plots corresponding to all six protein-ligand complexes identified in the preceding analysis. Concerning the protein target BRAC2, both Bayogenin and Olaparib achieved equilibrium at 25 ns, after which both plots exhibited stability. Minor fluctuations were recorded at specific time intervals of 85.4 ns, 108 ns, 130 ns, and 167 ns within the BRAC2-Bayogenin complex, whereas fluctuations at 127 ns and 115 ns were observed in the BRAC2-Olaparib complex, respectively. Both complexes demonstrated RMSD values within the range of (0.1–0.25) nm, which is within acceptable parameters. Furthermore, the average RMSD values for the BRAC2-Bayogenin and BRAC2-Olaparib complexes were determined to be 0.1466 ± 0.015569 nm and 0.1489 ± 0.016892 nm, respectively, throughout the 200 ns simulation period (Fig. 5a.). For the additional protein target BAX, the BAX-Bayogenin complex reached equilibrium at 8 ns, while the BAX-Olaparib complex attained equilibrium at 30 ns, with significant fluctuations discernible in both RMSD plots (Fig. 5b.). Notably, the primary fluctuations in the BAX-Bayogenin complex were recorded at 73 ns, 89 ns, and 107 ns, whereas in the BAX-Olaparib complex, they were noted at 148 ns. Moreover, two minor fluctuations were observed in the BAX-Olaparib complex at 112 ns and 123 ns. The average RMSD values were calculated to be 0.1654 ± 0.031052 nm and 0.1863 ± 0.035321 nm for the BAX-Bayogenin and BAX-Olaparib complexes, respectively. Overall, the RMSD trajectory of the BAX-Bayogenin complex exhibited comparatively greater stability than that of the BAX-Olaparib complex over the 200 ns period. The range of RMSD values was found to be (0.1–0.3) nm for the BAX-Bayogenin complex and (0.1–0.4) nm for the BAX-Olaparib complex. In the context of the PALB2 protein target, both the PALB2-Bayogenin complex and the PALB2-Olaparib complex reached equilibration at 3 ns. The PALB2-Bayogenin complex presented minor fluctuations at 112 ns, 145 ns, 164 ns, and 188 ns, while in the PALB2-Olaparib complex, fluctuations were observed at 108 ns and 122 ns. The range of RMSD values was determined to be (0.1–0.2) nm for both complexes, with average RMSD values recorded at 0.1377 ± 0.019289 nm and 0.1277 nm for the PALB2-Bayogenin and PALB2-Olaparib complexes, respectively (Fig. 5c.). A comparative analysis of the RMSD plots for all six protein-ligand complexes reveals that both Bayogenin and Olaparib exhibited relative stability in the context of RMSD evaluation.
Fig. 5
Representation of RMSD plot for (a) BRCA2-Bayogenin (black) and BRCA2-Olaparib (red), (b) BAX-Bayogenin (black) and BAX-Olaparib (red), (c)PALB2-Bayogenin (black) and PALB2-Olaparib (red), and representation of RMSF plot for (d) BRCA2-Bayogenin (black) and BRCA2-Olaparib (red), (e) BAX-Bayogenin (black) and BAX-Olaparib (red), (f) PALB2-Bayogenin (black) and PALB2-Olaparib (red)
RMSF trajectory analysisThe Root Mean Square Fluctuation (RSMF), representing the average structural deviation over time for each protein residue commencing from its initial reference position, was computed to evaluate the backbone stability of all six protein-ligand complexes during the simulation. Generally, diminished RMSF values are indicative of reduced individual flexibilities of each residue and enhanced stability of the protein-ligand complex. For the protein target BRAC2, the residues analyzed ranged from 850 to 1186. Both the BRAC2-Bayogenin and BRAC2-Olaparib complexes exhibited analogous patterns of fluctuating residues. Notably, residues 905, 998, 1079, 1090, and 1156 emerged as the principal fluctuating residues. The average RMSF values were determined to be 0.1277 ± 0.063926 nm and 0.1151 ± 0.046421 nm for the BRAC2-Bayogenin and BRAC2-Olaparib complexes, respectively (Fig. 5d.). In the case of the BAX-Bayogenin and BAX-Olaparib complexes, the residues included in the analysis spanned from 1 to 210, as per the crystal structure of the BAX protein. Numerous fluctuating residues were identified within this protein in association with both Bayogenin and Olaparib ligand molecules, which indicated comparatively lower stability in both the BAX-Bayogenin and BAX-Olaparib complexes relative to the other target protein. Average RMSF values of 0.1330 ± 0.054297 nm and 0.1532 ± 0.055607 nm underscored the relative stability of the BAX-Bayogenin complex over the BAX-Olaparib complex (Fig. 5e). Among the other protein-ligand complexes, both Bayogenin and Olaparib displayed reduced fluctuation in the RMSF plot for the protein target PALB2. The residues deemed relevant for this study were those ranging from 850 to 1185 according to the crystal structure of the PALB2 protein. Minor fluctuations were observed at residues 901 and 998 in both the PALB2-Bayogenin and PALB2-Olaparib complexes, displaying similar patterns and average RMSF values of 0.1431 ± 0.126726 nm and 0.1181 ± 0.062665 nm, respectively (Fig. 5f.). A comparative analysis of the RMSF plots across all six protein-ligand complexes reveals that both Bayogenin and Olaparib exhibited analogous patterns of fluctuations and stability across all three proteins.
Rg trajectory analysisThe radius of gyration (Rg) quantifies the extent of compactness within the protein-ligand complex. Generally, diminished Rg values are indicative of enhanced compactness and tighter folding of the protein structure. In the present investigation, Rg was computed to assess the impact of drug binding on the compactness of the corresponding proteins. Throughout the simulation for the BRCA2-Bayogenin and BRCA2-Olaparib complexes, Rg values were observed to fall within the range of (2-2.1) nm, with average Rg values calculated at 2.0687 ± 0.064942 nm and 2.0998 ± 0.008567 nm, respectively. For the BAX-Bayogenin and BAX-Olaparib complexes, Rg values ranged from (2.42–2.55) nm, with average values determined at 2.4761 ± 0.017794 nm and 2.4799 ± 0.016624 nm throughout the 200 ns trajectories. Similarly, the Rg range for the PALB2-Bayogenin and PALB2-Olaparib complexes was identified within (1.93–2.055) nm, yielding average Rg values of 1.9712 ± 0.016055 nm and 1.9537 ± 0.008912 nm, respectively. A comparative analysis of the Rg profiles for Bayogenin and Olaparib across the three protein targets revealed that both compounds exhibited comparable compactness concerning the BAX protein, thereby indicating greater stability of the BAX-Bayogenin complex in contrast to the other complexes (Fig. 6).
Fig. 6
Representation of Rg plot for (a)BRCA2-Bayogenin (black) and BRCA2-Olaparib (red), (b) BAX-Bayogenin (black) and BAX-Olaparib (red), (c)PALB2-Bayogenin (black) and PALB2-Olaparib (red), and representation of SASA plot for (d)BRCA2-Bayogenin (black) and BRCA2-Olaparib (red), (e) BAX-Bayogenin (black) and BAX-Olaparib (red), (f) PALB2-Bayogenin (black) and PALB2-Olaparib (red)
SASA trajectory analysisSASA quantifies the accessible surface area of proteins that can interact with solvent molecules. In this investigation, the SASA values of all six protein-ligand complexes were computed and scrutinized over a simulation duration of 200 ns. The trend observed across all six protein-ligand systems was remarkably similar; however, their mean SASA values exhibited slight variations. The calculated average SASA values were determined to be -24.95 ± 5.579925 nm², 27.75 ± 5.397654 nm², -75.87 ± 5.196412 nm², -74.87 ± 4.765381 nm², -19.86 ± 5.631884 nm², and − 13.75 ± 5.166198 nm² for the complexes BRCA2-Bayogenin, BRCA2-Olaparib, BAX-Bayogenin, BAX-Olaparib, PALB2-Bayogenin, and PALB2-Olaparib, respectively. Upon comparison of the six protein-ligand complexes, it is apparent that the average SASA value for Bayogenin in the BRCA2 complex was lower than that for Olaparib, indicating greater stability of the BRCA2-Bayogenin complex relative to the BRCA2-Olaparib complex in (Fig. 6.).
H-Bond analysisHydrogen bonding within a protein-ligand complex constitutes a fundamental determinant contributing to the stability of said complex. An increased quantity of hydrogen bonds correlates positively with enhanced stability of the protein-ligand complex. In the present investigation, the cumulative number of hydrogen bonds throughout 200 nanoseconds of simulation was systematically evaluated across all six protein-ligand systems. Within the BRCA2-Bayogenin and BRCA2-Olaparib complexes, three and four hydrogen bonds, respectively, were recorded throughout the simulation interval. Similarly, in the BAX-Bayogenin and BAX-Olaparib complexes, two and three hydrogen bonds were documented during the simulation period. Likewise, in the PALB2-Bayogenin and PALB2-Olaparib complexes, three and four hydrogen bonds were predominantly observed. Notably, it is important to highlight that across all three protein targets, Bayogenin exhibited the highest count of hydrogen bonds, ranging from four to six, while Olaparib presented a maximum of seven hydrogen bonds (Fig. 7.). From the analysis of hydrogen bonds, it becomes evident that while Bayogenin does not surpass Olaparib in total hydrogen bond formation, the relatively minor differences observed with protein targets such as BRCA2 underscore the significance of Bayogenin with the protein targets BRCA2, BAX, and PALB2.
Fig. 7
Representation of H-bond and PCA, (a) BRCA2-Bayogenin (black) and BRCA2-Olaparib (red), (b) BAX-Bayogenin (black) and BAX-Olaparib (red), (c) PALB2-Bayogenin (black) and PALB2-Olaparib (red)
Principal component analysis (PCA) and free energy landscape (FEL)All six protein-ligand complexes underwent a thorough examination utilizing Principal Component Analysis (PCA) and Free Energy Landscape (FEL) plots. The PCA plots facilitate the estimation of the overall fluctuations of the Cα atom, which enables the diagonalization of covariance matrices. A comprehensive analysis of the 200 ns trajectory was conducted for Bayogenin and Olaparib across all three protein targets, namely BRCA2, BAX, and PALB2. The accompanying (Fig. S1.). illustrates the PCA plot for all six protein-ligand systems. Within a PCA plot, a reduction in conformational space correlates with diminished overall motion of the protein, thereby indicating enhanced stability of the protein-ligand system. Olaparib exhibited a reduced conformational space across all three protein targets; however, Bayogenin demonstrated a comparable conformational space to Olaparib specifically on the BAX target, thereby underscoring the significance of Bayogenin for the BAX protein as deduced from the PCA analysis.
In addition to PCA, the FEL plot serves as a pertinent parameter for the assessment of the stability of the protein-ligand system. In the context of a FEL plot, singular low-energy basins (depicted as deep blue or dark regions) are indicative of energetically favorable conformations and signify a higher stability of the protein-ligand system. As observed in (Fig. S2), Bayogenin on BRCA2 exhibited comparatively narrower low-energy basins relative to Olaparib, thereby highlighting the superior stability of Bayogenin over Olaparib concerning the BRCA2 target. Both Bayogenin and Olaparib yielded comparable results concerning low-energy basins and stability on PALB2. Conversely, in the case of the BAX protein, Olaparib was found to exhibit greater stability than Bayogenin.
In summary, after comparing all the plots such as RMSD, RMSF, Rg, SASA, H-bond analysis for BRCA2-Bayogenin, BRCA2-Olaparib, BAX-Bayogenin, BAX-Olaparib, PALB2-Bayogenin, and PALB2-Olaparib, it was clear that both Bayogenin and Olaparib performed better on BRCA2 target than the BAX and PALB2 target. To further confirm the stability of the protein-ligand complex of Bayogenin and Olaparib on the BRCA2 targets we extended the simulation to 350 ns for more reliable and significant results. The outcome of the extended MDS revealed that Bayogenin performed better than Olaparib on the BRCA2 protein target (Fig. 8.)
Fig. 8
Representation of RMSF plot of BRCA2-Bayogenin (black) and BRCA2-Olaparib (red) for extended 350 ns MDS
MMPBSAMMPBSA was performed for the protein-ligand complex BRCA2-Bayogenin and BRCA2-Olaparib to further analyze the stability using the gmx_MMPBSA tool. In general, more negative is the total free binding energy of a protein-ligand complex, the more is its stability. The outcome from the MMPBSA showed that Bayogenin exhibited higher negative binding energy (-31.59 ± 5.47 kcal/mol) than Olaparib (-25.82 ± 3.09 kcal/mol) on BRAC2 protein. The total free binding energy revealed higher stability of Bayogenin than Olaparib. Table 3 shows detailed information about different forms of binding energy.
Table 3 MMPBSA calculations, where: ΔVDWAALS: Van der Waals contribution, ΔEEL: electrostatic energy, ΔEGB: Polar solvation free energy, ΔESURF: nonpolar solvation free energy, ΔGGAS: gas face free energy, ΔGSOLV: solvation energy, ΔTOTAL: total calculated free binding energy i.e. ΔGBinding (all units are reported in kcal/mol)
Comments (0)