Cell culture
The human cancer cell lines SNU449 (human hepatocellular carcinoma), SNU398 (human hepatocellular carcinoma), and A549 (human lung carcinoma) were purchased from the Korean Cell Line Bank, and 293A, a subclone of the human embryonic kidney 293 cell line, was purchased from Invitrogen (Carlsbad, CA, USA). All cells except SNU449 were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (HyClone Laboratories, Logan, UT, USA) at 37 °C in a humidified atmosphere containing 5% CO2; SNU449 was cultured in RPMI-1640 under the same conditions.
Reagents
Antibodies against β-actin (Cat#sc-47778), GAPDH (Cat#sc-365062), phospho-SAPK/JNK (Cat#sc-6254), c-Met (Cat#sc-8057), HSP27 (Cat#sc-13132), and GRP78 (Cat#sc-166490) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against phospho-Akt (Ser473) (Cat#9271 s), JNK (Cat#9252 s), phospho-JNK (Cat#9251 s), p38 (Cat#9212 s), phospho-p38 (Cat#9216 s), phospho-ERK(Cat#9101 s) were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies against GRP78 (Cat#11587-1-AP) and CD44(Cat#15675-1-AP) were purchased from Proteintech (Rosemont, IL, USA). An antibody against phospho-IRE1α (Cat#PA1-16927) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). An antibody against phospho-IGF1R (Tyr1161/Tyr1165/Tyr1166) (Cat#PA5-118536) was purchased from Sigma‒Aldrich (St. Louis, MO, USA). Sorafenib (Cat# SML2653) and MG132 (Cat#474787) were purchased from Sigma‒Aldrich, and GRP78 siRNA (Cat#sc-29338) was purchased from Santa Cruz Biotechnology. AEBSF (Cat#A-540) was purchased from Gold Biotechnology (St Louis, MO, USA). Antibodies against secondary Alexa Fluor 488 (Cat#A-11008) and Alexa Fluor 647 (Cat#A-31571) were purchased from Invitrogen (Carlsbad, CA, USA). MKC-3946 (Cat#A-12508) was purchased from AdooQ Bioscience (Irvine, CA, USA). TRIzol™ (Cat#15596018) was purchased from Life Technologies (Carlsbad, CA, USA). All other chemicals were purchased from Sigma‒Aldrich (St. Louis, MO, USA).
Construction of GRP78 (HSPA5) short hairpin RNAs (shRNAs)
First, we designed the target sequence as previously reported—a 2-base shift to the right—5’-GGTTACCCATGCAGTTAC-3’ and made two strands for annealing to produce shRNA. The loop sequence was 5’-TCTC’-3’, and the poly A sequence was 5’-TTTTA-3’. The top strand was 5’-GATCC GGTTACCCATGCAGTTGTTAC TCTC GTAACAACTGCATGGGTAACC TTTTA-3’; and the bottom strand was 5’-AGCTT AAAA GGTTACCCATGCAGTTGTTAC GAGA GTAACAACTGCATGGGTAACC G-3’. To construct more potent human GRP78 shRNAs, we screened five candidate sequences via an algorithm developed by Genolution Pharmaceuticals, Inc. (Seoul, Korea). The GRP78 shRNA candidates for the target sequence were as follows:
(1) 5’-CAAAGACGCUGGAACUAUUGCUCUCGCAAUAGUUCCAGCGUCUUUG UU-3’; (2) 5’-CUAUUGCUGGCCUAAAUGUUAUCUC UAACAUUUAGGCCAGCAAUAGUU-3’; (3) 5’-GCUUAUGGCCUGGAUAAGAGGUCUCCCUCUUAUCCAGGCCAUAAGCUU-3’; (4) 5’-GGUCUAUGAAGGUGAAAGACCUCUC GGUCUUUCACCUUCAUAGACC UU-3’; and (5) 5’-CCAAGAUGCUGACAUUGAAGAUCUC UCUUCAAUGUCAGCAUCUUGGUU-3’. The negative control shRNA provided by Genolution was 5’-CCUCGUGCCGUUCCAUCAGGUAGUCUCCUACCUGAUGGAACGGCACGAGG UU-3’. The final target sequence selected was 5’-CCAAGAUGCUGACAUUGAAGA-3’, which was derived from GRP78 shRNA 5. For expression of human GRP78 shRNA in an adenovirus vector, the top strand sequence was 5’-GATCCCCAAGATGCTGACATTGAAGATCTCTCTCTTCAATGTCAGCATCTTGGTTTTTTA-3’, and the bottom strand sequence was 5’-AGCTTAAAAAA CCAAGATGCTGACATTGAAGA GAGAGA TCTTCAATGTCAGCATCTTGGG-3’. The loop sequence was 5’-TCTCTC-3’, and the poly A sequence was 5’-TTTTTTA-3’. The top and bottom strands were annealed for subcloning into BamHI–HindIII-digested pSP72-ΔE3-H1-shDaxx to generate pSP72-ΔE3-H1-shGRP78.
Construction of vimentin shRNAs
Of the two validated target sequences (5’-GAAGAATGGTACAAATCCAAGTTTG-3’, 5’-TGAAGCTGCTAACTACCAAGACACT-3’) available, the second was selected by western blotting after the shuttle vector (pSP72-ΔE3-shvimentin 1st, 2nd) was transfected into 293A cells. The selected top strand sequence was 5’-GATCC TGAAGCTGCTAACTACCAAGACACT TCTC AGTGTCTTGGTAGTTAGCAGCTTCA TTTT A-3’, and the bottom strand sequence was 5’-AGCTT AAAA TGAAGCTGCTAACTACCAAGACACT GAGA AGTGTCTTGGTAGTTAGCAGCTTCA G-3’. The top and bottom strands were subsequently annealed for subcloning into the BamHI-HindIII-digested pSP72-ΔE3-U6-siGFP to make pSP72-ΔE3-U6-shvimentin (2nd).
Construction of CD44 standard shRNAs
To construct the most potent human CD44 shRNA, we screened four candidate shRNA sequences after subcloning them into BamHI-HindIII-digested pSP72-ΔE3-U6-siGFP to generate pSP72-ΔE3-U6-shCD44s. (1) top strand 5’-GATCC GCAGCACTTCAGGAGGTTACA TCTCTC TGTAACCTCCTGAAGTGCTGC TTTTA -3’ and bottom strand 5’-AGCTT AAAA GCAGCACTTCAGGAGGTTACA GAGAGA TGTAACCTCCTGAAGTGCTGC G-3’; (2) top strand 5’-GATCC GCACTTCAGGAGGTTACATCT TCTCTC AGATGTAACCTCCTGAAGTGC TTTTA -3’ and bottom strand 5’-AGCTT AAAA GCACTTCAGGAGGAGGTTACATCT GAGAGA AGATGTAACCTCCTGAAGTGC G-3’; (3) top strand 5’-GATCC GCTTTGATTCTTGCAGTCAAC TCTCTC GTTGACTGCAAGAATCAACTC TTTTA -3’ and bottom strand 5’- AGCTT AAAA GCTTTGATTCTTGCAGTCAAC GAGAGA GTTGACTGCAAGAATCAAAGC G-3’; (4) top strand 5’-GATCC GGACCAATTACCATAACTATT TCTCTCT AATAGTTATGGTAATTGGTCC TTTTA- 3’ and bottom strand 5’-AGCTT AAAA GGACCAATTACCATAACTATT GAGAGA AATAGTTATGGTAATTGGTCC G-3’. The 4th target sequence (GGACCAATTACCATAACTATT) originated from the 2nd siRNA of CD44 in Hu et al. [28]. Then, the 4th was selected by real-time PCR and western blotting after transfection.
Construction of HIF-1α shRNAs
To construct the most potent human HIF-1α shRNA, we screened six candidate shRNA sequences after subcloning into BamHI-HindIII-digested pSP72-ΔE3-U6-siGFP to generate pSP72-ΔE3-U6-shHIF-1α. (1) top strand 5’-GATCC GCCGAGGAAGAACTATGAACA TCTCTC TGTTCATAGTTCTTCCTCGGC TTTTTT A-3’ and bottom strand 5’-AGCTT AAAAAA GCCGAGGAAGAACTATGAACA GAGAGA TGTTCATAGTTCTTCCTCGGC G-3’; (2) top strand 5’-GATCC GCTGATTTGTGAACCCATTCC TCTCTC GGAATGGGTTCACAAATCAGC TTTTTT A-3’ and bottom strand 5’-AGCTT AAAAAA GCTGATTTGTGAACCCATTCC GAGAGA GGAATGGGTTCACAAATCAGC G-3’; (3) top strand 5’-GATCC GCTGGAGACACAATCATATCT TCTCTC AGATATGATTGTCTCCAGC TTTTTT A-3’ and bottom strand 5’-AGCTT AAAAAA GCTGGAGACACAATCATATCT GAGAGA AGATATGATTGTGTCTCCAGC G-3’; (4) top strand 5’-GATCC GCAGCTACTACATCACTTTCT TCTCTC AGAAAGTGATGTAGTAGCTGC TTTTTT A-3’ and bottom strand 5’-AGCTT AAAAAA GCAGCTACTACATCACTTTCT GAGAGA AGAAAGTGATGTAGTAGCTGC G-3’; (5) top strand 5’-GATCC GCTACTACATCACTTTCTTGG TCTCTC CCAAGAAAGTGATGTAGTAGC TTTTTT A-3’ and bottom strand 5’-AGCTT AAAAAA GCTACTACATCACTTTCTTGG GAGAGA CCAAGAAAGTGATGTAGTAGC G-3’; (6) top strand 5’-GATCC AAGAAACTACTAGTGCCACATCA TCTCTC TGATGTGGCACTAGTAGTTTCTT TTTTTT A-3’ and bottom strand 5’-AGCTT AAAAAA AAGAAACTACTAGTGCCACATCA GAGAGA TGATGTGGCACTAGTAGTTTCTT G-3’. The 6th target sequence (AAGAAACTACTAGTGCCACATCA) was originated from the 5th target sequence of HIF1α in Mizuno et al. [29]. Then, the 6th was selected by real time PCR after transfection.
Construction of a defective adenovirus expressing U6-shTGF-β1-H1-shGRP78
pSP72-ΔE3-U6-shTGF-β1-H1-shGRP78 was constructed by inserting H1-shGRP78-Poly A into HindIII, followed by blunting/KpnI-digested pSP72-ΔE3-U6-shTGF-β1 after the digestion of pSP72-ΔE3-H1-shGRP78 with SphI, followed by blunting/KpnI. pSP72-ΔE3-U6-shTGF-β1-H1-shGRP78 was linearized by XmnI and then cotransformed into E. coli BJ5183, together with a SpeI-digested dl324-IX backbone for homologous recombination.
Construction of a defective adenovirus expressing H1-shHSP27-U6-shTGF-β1
pSP72-ΔE3-H1-shHSP27-U6-shTGF-β1 was constructed by inserting U6-shTGF-β1-Poly A into HindIII, followed by blunting/KpnI-digested pSP72-ΔE3-H1-shHSP27 after digestion of pSP72-ΔE3-U6-shTGF-β1 with SphI, followed by blunting/KpnI. pSP72-ΔE3-H1-shHSP27-U6-shTGF-β1 was linearized by XmnI and then cotransformed into E. coli BJ5183, together with a SpeI-digested dl324-IX backbone for homologous recombination.
Construction of a defective adenovirus expressing U6-shTGF-β1-U6-shvimentin
pSP72-ΔE3-U6-shTGF-β1-U6-shvimentin was constructed by inserting U6-shvimentin-Poly A into Hind III, followed by blunting/KpnI-digested pSP72-ΔE3-U6-shTGF-β1 after the digestion of pSP72-ΔE3-U6-shvimentin with SphI, followed by blunting/KpnI. pSP72-ΔE3-U6-shTGF-β1-U6-shvimentin was linearized by XmnI and then cotransformed into E. coli BJ5183, together with a SpeI-digested dl324-IX backbone for homologous recombination.
Construction of an oncolytic adenovirus expressing HSP27 and TGF-β1 shRNAs
The adenoviral E3 region shuttle vector pSP72-ΔE3-H1-shHSP27-U6-shTGF-β1 was linearized via XmnI digestion, and the adenoviral backbone vector dl324-BstBI was linearized via SpeI digestion. The two linearized vectors were subsequently cotransformed into E. coli BJ5183 competent cells for the first homologous recombination. A second homologous recombination was subsequently performed using dl324-BstBI-H1-shHSP27-U6-shTGF-β1 and the adenoviral shuttle vector pVAX1-3484, which contains an E1 region without E1B55K, to generate an E1B55K-deleted oncolytic adenovirus harboring HSP27 and TGF-β1 shRNAs. The final adenoviral construct, dl324-3484-H1-shHSP27-U6-shTGF-β1, was digested with PacI and transfected into 293A cells to generate a tumor-selective replicative adenovirus. The 293A cells were reinfected with the generated adenovirus, and this procedure was repeated for viral amplification. The infectious titer of the adenovirus was determined by performing a limiting dilution assay in 293A cells after concentration and purification with CsCl gradients.
Construction of an oncolytic adenovirus expressing H1-shHSP27-U6-shTGF-β1-U6-shHIF1α (or U6-shTGF-β1-H1-shGRP78-U6-shHIF1α)
First, the adenoviral E3 region shuttle vector pSP72ΔE3-H1-shHSP27-U6-shTGF-β1-U6-shTGF-1α (or pSP72ΔE3-U6-shTGF-β1-H1-shGRP78-U6-shHIF-1α) was constructed by inserting U6-shHIF-1α-Poly A into HindIII followed by blunting/KpnI-digested pSP72-ΔE3-H1-shHSP27-U6-shTGF-β1 (or pSP72ΔE3-U6-shTGF-β1-H1-shGRP78) after the digestion of pSP72-ΔE3-U6-shHIF-1α with SphI followed by blunting/KpnI. pSP72-ΔE3-H1-shHSP27-U6-shTGF-β1-U6-shHIF-1α was linearized by XmnI and then cotransformed into E. coli BJ5183, together with a SpeI-digested dl324-IX backbone for the first homologous recombination. A second homologous recombination was performed using dl324-BstBI-H1-shHSP27-U6-shTGF-β1-U6-shHIF1α (or dl324-BstBI-U6-shTGF-β1-H1-shGRP78-U6-shHIF1α) and the adenoviral shuttle vector pVAX1-3484, which contains an E1 region without E1B55K, to generate an E1B55K-deleted oncolytic adenovirus harboring HSP27, TGF-β1, and HIF1α shRNAs. The final adenoviral construct, dl324-3484-H1-shHSP27-U6-shTGF-β1-U6-shHIF1α (or dl324-BstBI-U6-shTGF-β1-H1-shGRP78-U6-shHIF1α), was processed for viral production as described in the preceding sentence.
Establishment of sorafenib-resistant cells
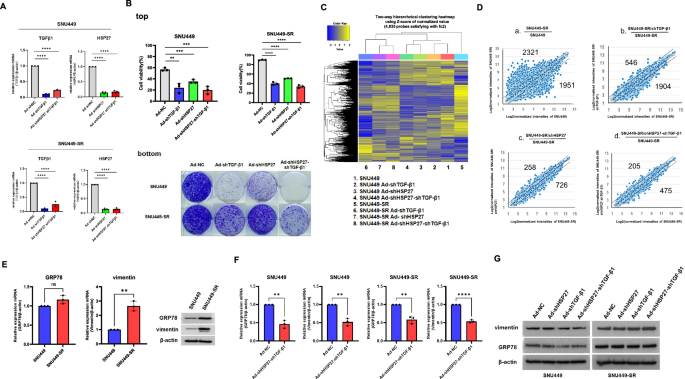
The IC50 of sorafenib in HCC cells was initially determined by incubating cells with different concentrations of sorafenib in 96-well plates, and cell viability was measured 24 h later via an MTS viability assay as described in Kang et al. [30]. Two HCC cell lines (SNU449 and SNU398) were cultured in 6-well plates at 1 × 104 cells/well and incubated with sorafenib at concentrations below the IC50 (10 μM and 5 μM, respectively). The sorafenib concentration was progressively increased by 0.5 μmol/L per week. After the IC50 values were reached (15 μM and 7.5 μM, respectively), two sorafenib-resistant HCC cell lines, termed SNU449-SR (less sensitive) and SNU398-SR (more sensitive), were obtained and were continuously maintained by culture in the presence of sorafenib (10 μM and 5 μM, respectively).
MTS assay
The CellTiter® 96 AQueous Assay Kit (Promega, Madison, WI, USA) is composed of solutions containing a novel tetrazolium compound (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; MTS) and an electron-coupling reagent (phenazine ethosulfate; PES). The MTS is bioreduced by cells into a formazan product that is soluble in tissue culture media. SNU449, SNU449-SR, SNU398, and SNU398-SR cells were cultured in a six-well plate with adenovirus infection (dl324-IX-ΔE3-U6-shNC, dl324-IX-ΔE3-U6-shTGF-β1, dl324-IX-ΔE3-H1-shHSP27, dl324-IX-ΔE3-H1-shGRP78, dl324-IX-ΔE3-U6-shvimentin, dl324-IX-ΔE3-H1-shHSP27-U6-TGF-β1, or dl324-IX-ΔE3-U6-TGF-β1-H1-shGRP78 at an MOI of 50 for 2 days. Then, 100 μL of trypsinized cell-containing supernatant from each well was transferred into a new well of a 96-well flat-bottom plate. The absorbance of formazan at 490 nm was measured directly from the 96-well assay plates without additional processing. The formazan concentration was directly proportional to the number of living cells in each culture.
Clonogenic assays
The cells were plated into six-well plates at a density of 1 × 105 cells/well. SNU449, SNU449-SR, SNU398, and SNU398-SR cells were subsequently cultured in a 6-well plate with adenovirus infection (dl324-IX-ΔE3-U6-shNC, dl324-IX-ΔE3-U6-shTGF-β1, dl324-IX-ΔE3-H1-shHSP27, dl324-IX-ΔE3-H1-shGRP78, dl324-IX-ΔE3-U6-shvimentin, dl324-IX-ΔE3-H1-shHSP27-U6-TGF-β1, or dl324-IX-ΔE3-U6-TGF-β1-H1-shGRP78) at an MOI of 50 for 2 days. The cells were then trypsinized and plated into six-well plates at densities of 5 × 103 or 1 × 104 cells/well. The cultures were monitored daily via microscopy. When the cells formed colonies, the surviving cells were fixed with 4% paraformaldehyde and stained with 0.5% crystal violet.
Immunoblot analysis
The cells were lysed in 1× Laemmli lysis buffer (62.5 mM Tris, pH 6.8; 2% sodium dodecyl sulfate; 10% glycerol; 0.02% bromophenol blue). Proteins were separated by SDS‒PAGE and electrotransferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After each membrane (containing 5% nonfat dry milk in TBS-Tween-20 (0.1%, v/v) was blocked at room temperature for 1 h), it was incubated with primary antibody (diluted according to the manufacturer’s instructions) for durations ranging from 2 h to overnight, depending on the antibody. Horseradish peroxidase-conjugated anti-mouse, anti-rabbit or IgG was used as a secondary antibody. Immunoreactive proteins were visualized via chemiluminescence (Thermo Fisher Scientific, Waltham, MA, USA).
Real-time PCR
SNU449, SNU449-SR, SNU398, or SNU398-SR cells were infected with a defective adenovirus expressing human TGF-β1 shRNA, GRP78 shRNA, TGF-β1/GRP78 shRNAs or scrambled shRNA. After 2 days of infection, the cells were lysed with TRIzol™ reagent (Life Technologies), and total RNA was isolated via chloroform extraction. The RNA concentration was determined via a Nanodrop™ 2000 (Thermo Scientific, Fremont, CA, USA). A real-time polymerase chain reaction (PCR) assay was performed using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, Waltham, MA, USA). The reaction mixture comprised an RT enzyme mixture, reverse transcription (RT)-PCR mixture, forward primer, reverse primer, RNA template, and nuclease-free water. Human GRP78 was amplified with the forward primer 5’-CTTGCCGTTCAAGGTGGTTG-3’ and the reverse primer 5’-GCATGGGTAACCTTCTTTCC-3’. Human β-actin was amplified with the forward primer 5’-ACTCTTCCAGCCTTCCTTC-3’ and the reverse primer 5’-ATCTCCTTCTGCATCCTGTC-3’.
Microarray analyses
The cDNA microarray assay was performed using total RNA isolated from two cancer cell lines 48 h after infection with adenoviruses bearing TGF-β1 shRNA, HSP27 shRNA, GRP78 shRNA, both TGF-β1 shRNA and HSP27 shRNA, or both TGF-β1 shRNA and GRP78 shRNA. Total RNA was extracted and purified via the RNeasy Plus Mini Kit (Qiagen, 74134) according to the manufacturer’s instructions. The results were summarized and normalized via Affymetrix Power Tools (APT). These data were exported for differentially expressed gene (DEG) analysis. The statistical significance of the expression data was determined via the fold-change magnitude. Hierarchical cluster analysis was performed for the DEG set using complete linkage and Euclidean distance as measures of similarity. All the data analyses and visualizations of the DEGs were conducted by Macrogen, Inc. (Korea) via R 3.3.2.
Measurement of cell-surface GRP78 by biotinylation and avidin pulldown
The cell surface expression of the GRP78 protein was measured via a biotinylation and avidin pulldown method as described previously [31]. After the addition of sorafenib for the indicated times, the cells were washed with cold PBS three times. Then, 1 mg/mL EZ-Link Sulfo-NHS-SS-Biotin (Thermo Scientific, Waltham, MA) in PBS was added, and the cells were gently shaken at room temperature for 30 min. To stop the biotinylation reaction, the biotin solution was removed, and the cells were rinsed 3 times with quenching buffer (cold PBS containing 100 mM glycine). Then, the cells were subjected to a radioimmunoprecipitation assay (RIPA). Part of the whole-cell lysate was saved for immunoblotting to measure the total levels of the target proteins. To purify the surface proteins, the remaining lysate was mixed with NeutrAvidin™ agarose resin (Thermo Scientific) at room temperature for 1 h, and the resulting mixture was washed with RIPA buffer and centrifuged at 3000 × g for 1 min 6 times. The cell-surface proteins were released by the addition of 50 μL of 2× SDS‒PAGE sample buffer, followed by heating at 95 °C for 5 min and centrifugation at 6000 × g for 5 min to collect the supernatant to perform immunoblot analyses for the detection of surface proteins.
Confocal microscopy
HCC cell lines (SNU449 and SNU398) were cultured in confocal plates at 1 × 105 cells/well and incubated with sorafenib at concentrations below the IC50 (10 μM and 5 μM, respectively). Then, the cells were fixed in 4% PFA for 15 min at room temperature and incubated with primary antibodies against CD44 (Proteintech 15675-1), HSP27 (Santacruz sc-13132), CD44 (Proteintech 15675-1) and GRP78 (Santacruz sc-166490) at room temperature for 2 h and with Alexa Fluor 488-conjugated anti-rabbit IgG (Invitrogen A-11008) or Alexa Fluor 647-conjugated anti-mouse IgG (Invitrogen A-31571) at room temperature for 1 h. Nuclei were stained with DAPI (Invitrogen D1306) at room temperature for 5 min. Fluorescence images were taken via an LSM700 (Carl Zeiss) confocal microscope.
Capturing Z-stack confocal images
MemBrite® Fix Cell Surface Staining Kits are reactive fluorescent dye stains that react with cell surface proteins (biotium, 30094-T). SNU449 and SNU398 cells were seeded in confocal dishes and treated with sorafenib at concentrations below the IC50 (10 μM and 5 μM, respectively). Then, MemBrite® 543/560 cell surface prestaining solution was added, and the mixture was incubated at 37 °C for 5 min. After that, the MemBrite® solution was incubated at 4 °C for 30 min. The cells were subsequently fixed in the same way as mentioned above. Finally, fluorescence images were taken via an LSM700 (Carl Zeiss) confocal microscope with Z-stack settings.
Establishment of the SNU398/CD44 cell line
A stable SNU398 cell line expressing CD44 was selected with puromycin. SNU398 cells were infected with retrovirus produced by transfecting the retroviral pBabe-puro-CD44s vector (Addgene #19127) into Platinum-A packaging cells (Cell Biolabs, United States), and then, a selection process was performed via puromycin (3 μg/ml). After several clones were selected, CD44 expression was confirmed through western blotting, after which the final clone was selected. As a negative control, SNU398/NC cells were also produced via the pBabe puro empty vector.
Animal study
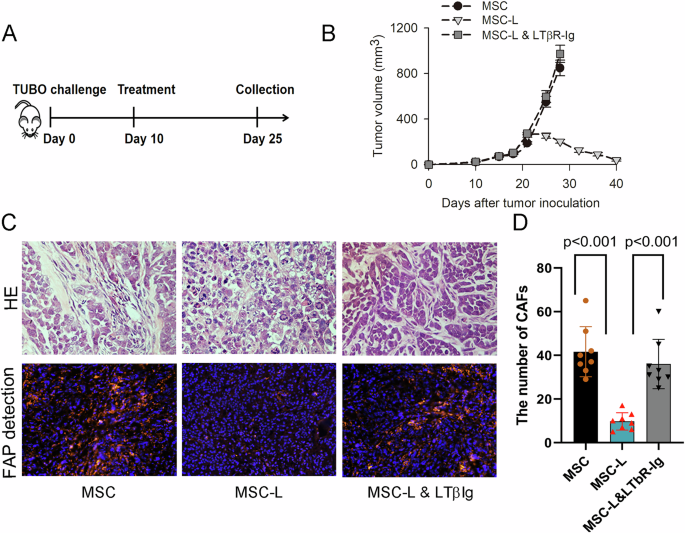
An animal study was performed in compliance with the ARRIVE guidelines as described below. For the experiment, 5-week-old male BALB/c athymic nude mice weighing approximately 20 g were obtained from OrientBio (Seongnam, Korea). All the mice were housed in individually ventilated cages at 21\(\pm\)2 °C and 50\(\pm\)10% humidity while undergoing a 1-week acclimation period with free access to food (PicoLab Rodent diet 20 5053) and reverse osmosis-purified water. The maximum caging density was five mice per cage, and autoclaved aspen chips were used as bedding. To generate a xenograft tumor model, 2 × \(^\) SNU398-SR cancer cells in Matrigel™ were injected subcutaneously into the flank region of the mice. When the tumors reached an average size of 70−120 mm³, 5 × 108 plaque-forming units (PFUs) of each adenovirus diluted in 50 μL of PBS or PBS alone were injected intratumorally into the nude mice. Mice with similar average and standard deviation values of tumor size were separated into groups. The adenoviruses used for injection were Ad-NC, Ad-shHSP27-shTGF-β1, or Ad-shTGF-β1-shGRP78. Intratumoral injection was repeated twice every other day. The arrows indicate when the oncolytic adenovirus was administered. The error bars represent the means ± SDs from 3 to 5 mice per group: *P < 0.05. Repression of tumor growth was assessed by measuring the length (L) and width (W) of the tumors. The tumor volume was calculated via the following formula: volume (mm³) = 0.52 × L × W2. Although no formal statistical power analysis was performed, the sample size (n = 3–5 per group) was determined based on prior experience with xenograft models and aimed to balance biological relevance and ethical considerations. No formal randomization method was used. However, mice with comparable baseline tumor volumes were grouped together to minimize allocation bias. No blinding was performed during the experiment or outcome assessment. Tumor volumes were measured using standardized caliper-based methods to reduce observer bias. All animal experiments were conducted in accordance with the institutional guidelines for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee (IACUC) of Yonsei University (approval number: 2022-0111).
Statistical analyses
The results are presented as the mean ± standard deviation (SD) with error bars representing the SD. Differences between groups were examined via Student’s t tests or two-way analysis of variance (ANOVA). P values were calculated via GraphPad Prism 10, and p < 0.05 was considered statistically significant.

Comments (0)