
Remember me
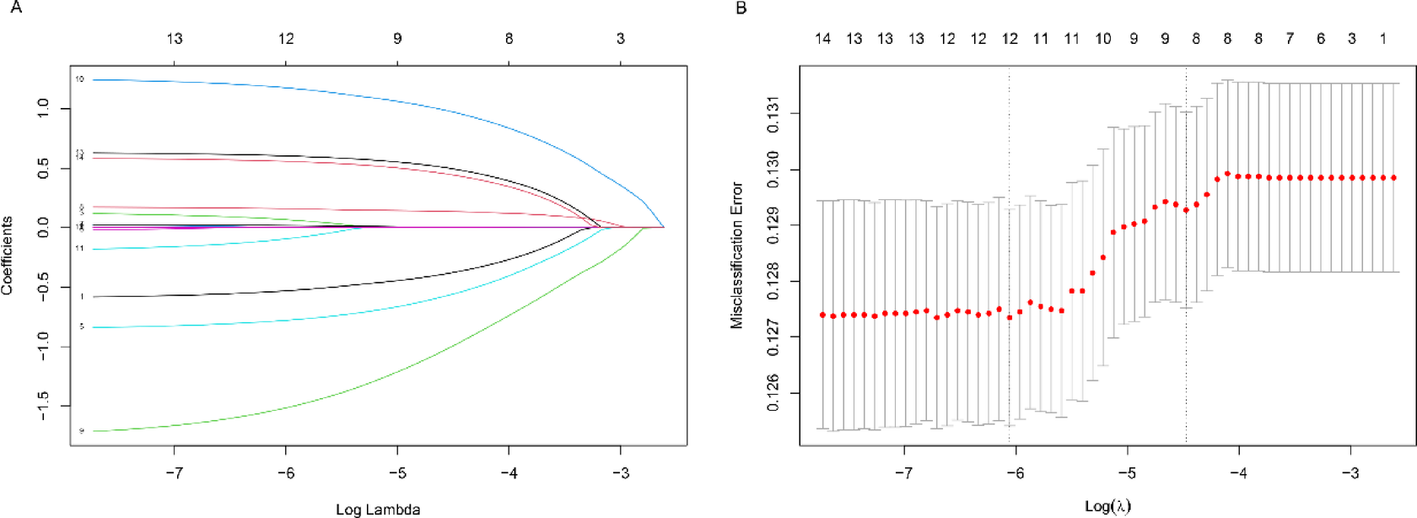
A consensus clustering approach was performed to group patients with osteosarcoma in the dataset into subgroups based on 139 coagulation-related genes. When K = 2, optimal cluster stability was found (Fig. 1C-E). Cluster 1 consisted of 90 patients, while Cluster 2 consisted of 49 individuals. CRG expression levels in both subtypes were displayed using a heat map (Fig. 1C), and substantial expression variations were seen between clusters 1 and 2. Furthermore, patients in cluster 1 had a higher overall survival rate than those in cluster 2 (P < 0.05; Fig. 1F). These findings imply that CRG divides osteosarcoma patients into two molecular subgroups with varying overall survival.
Fig. 1
(A) PCA before removal of inter-batch differences. (B) PCA after removal of inter-batch differences. Consensus cluster. (C–E) K = 2 was identified the optimal value for consensus clustering. (F) survival curve of the patients in the two subgroups
3.2 Two molecular subtypes’ immune statusFirst, we reviewed the immune microenvironment of the two tumor groups and concluded that the immune and stromal scores of cluster1 were much higher than those of cluster 2, however, cluster 2 was much superior than cluster 1 in terms of tumor purity (Fig. 2A). We then analyzed the 2 clusters by ssGSEA, and among the 28 immune cells, cluster 1 was more expressed in most immune cells than cluster 2, such as Mast cells, Central memory CD8 T cells (Fig. 2B) and with the further study, we analyzed the immune cells in both groups by using TIMER, and among these six immune cells, only B cell was not dissimilar between the 2 clusters (P > 0.05), and cluster one was much upper than cluster two in all other immune cells (Fig. 2D). “CIBERSORT”, “QUANTISEQ”, “XCELL”, “EPIC” and “MCPCOUNTER” were worked to study the immune disparities between the two groups, and the heatmap presented that the expression of most immune cells was much higher in cluster 1 than in cluster 2 (Fig. 2 C). Subsequently, we analyzed the gaps in the expression of immune regulatory genes and immune checkpoint genes between the two groups, and we found that cluster 1 was more highly expressed than cluster 2 in both immune checkpoint and immune regulatory genes (Fig. 3A-B). We found that the expression of Human leukocyte antigen (HLA) gene was much higher in the first group than in the second group (Fig. 3D). Finally, comparison of cytolytic activity scores revealed that the first group exhibited significantly higher scores than the second group (Fig. 3C). Overall, the immune microenvironment and immune cells in cluster 1 were far better than those in cluster 2.
Fig. 2
Immune analyses in the two clustered subgroups. (A)ESTIMATE score, Stromal score, Immune score, tumor purity. (B) statistical analysis of ssGSEA. *p < 0.05; **p < 0.01; ***p < 0.001. (C) The heat map depicts the immunological assessment of the two immune profiles by seven immunization methods. (D) abundance of six immune filtrating cells evaluated by TIMER
Fig. 3
(A) Immune checkpoint genes. (B) Immunomodulatory genes. (C) Immunolytic cell score. (D) Differences in HLA family genes between the two groups Network
3.3 Analysis of differences between the two clusters and functional enrichmentWe performed differential analysis of gene expression between the two groups, and to investigate the differences in biological functions between the two groups, we performed KEGG, GO, and PPI network construction for these 137 differential genes, including 90 up-regulated genes as well as 47 down-regulated genes(Supplementary Fig. 1A). We found that the functions of these differential genes were mainly enriched in “extracellular matrix organization”, “Staphylococcus aureus infection” and "immunoglobulin mediated immuneresponse" Supplementary Fig. 1B-D.
3.4 Construction of prognostic typeLASSO-based regression analysis was applied to the genes, and the COX stepwise regression method was used to construct the prognostic model, and the final prognostic model was obtained based on 25 genes (Supplementary Fig. 2A-B; Supplementary Fig. 4A).
3.5 Evaluation of prognostic modelsThe sample could be divided into two subgroups according to high and low risk based on median risk values. The Kaplan-Meier survival curves between the two groups are shown below with a noteworthy difference in survival between the two (P < 0.01; Supplementary Fig. 1C). The survival status of the patients between the risk groups shows a significant increase in mortality with increasing risk factors can be seen (Supplementary Fig. 1E). In order to more accurately assess the accuracy of the prognostic model, the ROC curves of the prediction model at 1, 3 and 5 years were plotted in this study and the corresponding AUC values were calculated Supplementary Fig. 1D. The results showed that the AUC values of the prediction model were 0.824, 0.802 and 0.814 at 1, 3 and 5 years in the training set, respectively, which were all greater than 0.75, indicating that the model has a high prediction accuracy.
In the validation group dataset, we constructed COX regression models using the same variables, and survival analysis showed the same grave dissimilarity in survival rates between the high and low risk groups. The mortality rate of patients increased significantly with increasing risk scores (Supplementary Fig. 1 A,C). Based on the risk scores of the model, the ROC curves predicted the AUC values of the validation set patients at 1, 3 and 5 years, which were 0.934, 0.708 and 0.782, respectively, indicating that the prognostic model in the training set could also accurately forecast the survival of patients Supplementary Fig. 3B.
Subsequently, a multifactorial COX regression analysis was executed on the risk scores of the training set sample and clinical data of the patients, including patient age, patient gender, and presence of metastasis. The results showed that the patients’ cancer progression status and risk score were independent poor prognostic factors affecting the survival of patients with osteosarcoma. Patients without distant metastases had a better prognosis than those with distant metastases, with a risk ratio of 5.94 (95% CI 2.24–15.8) and a P value of less than 0.001(Supplementary Fig. 2 F).
In this study, a multifactorial COX regression analysis was performed on the risk scores of the validation set sample as well as on the clinical data of the patients. Both results were consistent, again demonstrating that patients’ cancer progression status and risk scores were independent risk factors affecting survival in patients with osteosarcoma. The wide applicability and stability of our prognostic model is more fully illustrated(Supplementary Fig. 3D).
Next, a prognostic Nomogram was constructed for this study based on the patient’s age, gender, metastatic status, and risk score of the prognostic model. The value of each variable can be represented by a line perpendicular to the horizontal axis, and the score of that variable is located at the point where the line intersects the coordinate axis. The total score value can be obtained by adding the scores corresponding to each variable. The graph demonstrates that factors such as age, male gender, tumor metastasis, and high-risk score significantly affect patient survival. The probability of survival at 1, 3 and 5 years for patients with osteosarcoma can be visually predicted based on the Nomogram (Supplementary Fig. 3E).
3.6 Pan-cancer analysis on ITIH1We investigated the expression statistics of 34 cancer kinds and discovered that ITIH1 was vastly expressed in some tumor samples and statistically different. For example, it was significantly upregulated in 5 tumors including GBM, KIPAN, KIRC, ALL and KICH (Supplementary Fig. 4C).
To further explore the relationship between ITIH1 and pan-cancer prognosis, we made a prognostic analysis of 44 cancer sorts. The results of overall survival (OS) showed that for GBMLGG, LGG, STES, KIPAN, and KIRC, the higher the expression of ITIH1, the lesser the survival rate of tumor patients (P < 0. 05; Supplementary Fig. 3D). We also plotted the survival curves of GBMLGG, LGG, STES and KIPAN (Supplementary Fig. 3B).
In GBM, ITIH1 high expression group had lower mutation rate in “ADGRV1” and “CACNA1S” than ITIH1 low expression group; in KIRC, ITIH1 high expression group had higher mutation rate in “BAP1” and “HMCN1” than ITIH1 low expression group. “BAP1” and “HMCN1” gene mutation rate was higher than that of ITIH1 low expression group; in LGG, ITIH1 high expression group had higher mutation rate in “BAP1”, “NOTCH1”, “TTN” and “BAP1” genes than ITIH1 low expression group, In LGG, the mutation rates of “BAP1”, “NOTCH1”, “SMARCA4” and “TTN” genes were higher in the ITIH1 high expression group than in the ITIH1 low expression group(Supplementary Fig. 4E). Supplementary Fig. 4F shows a lollipop plot of the highly mutated gene ITIH1 in various cancers.
Comments (0)