
Remember me
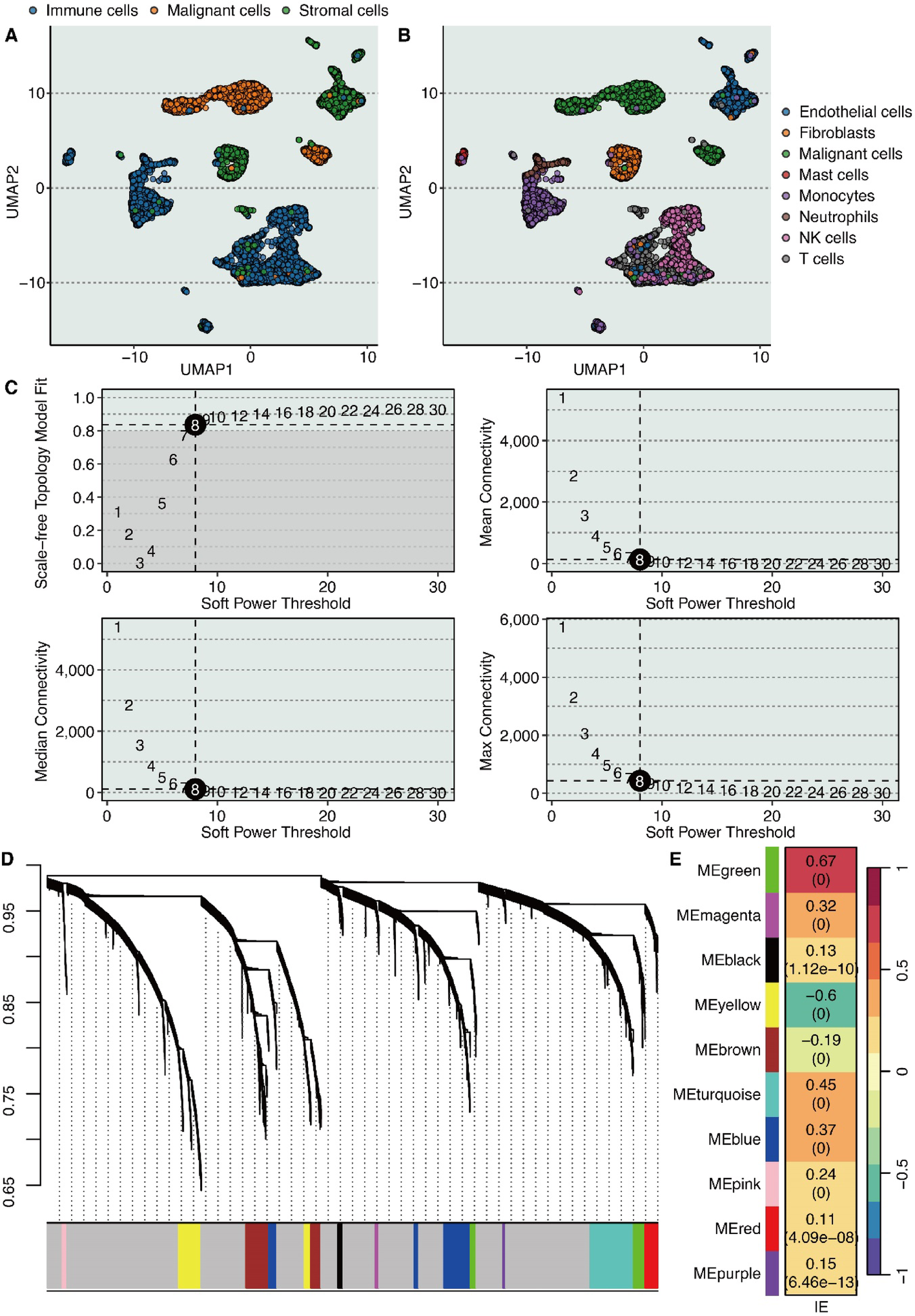
The TME of KIRC contains three major cell populations: immune cells, malignant cells, and stromal cells (Fig. 1A). Further analysis revealed additional cell types, including endothelial cells, fibroblasts, mast cells, monocytes, neutrophils, NK cells, and T cells (Fig. 1B). The hdWGCNA algorithm identified eight as the optimal soft threshold power (Fig. 1C). Gene modules derived from hdWGCNA are presented in Fig. 1D, with the green module showing the strongest association (correlation coefficient = 0.67) with the IE feature (Fig. 1E).
Fig. 1
Identification of IE-related gene module. A UMAP shows the major cell types in the tumor microenvironment of KIRC. B UMAP shows the minor cell types in the tumor microenvironment of KIRC. C The association between soft power threshold and scale-free topology model fit, mean connectivity, median connectivity, and max connectivity by hdWGCNA. D Waterfall plot shows the distribution of gene modules by hdWGCNA. E The correlation between the IE feature and gene modules
3.2 Development of IE patternsWe first analyzed IE-related genes using univariate Cox regression. Among these, 16 genes showed significant expression differences between tumor and normal tissues (Fig. 2A). Using PAM clustering, we identified two distinct IE patterns (Fig. 2B). KIRC samples clearly segregated into these two subgroups via principal component analysis (PCA) (Fig. 2C). Notably, patients of IE pattern 1 exhibited significantly longer OS compared to pattern 2 (Fig. 2D).
Fig. 2
Development of IE patterns. A The differential expression of 16 IE-related genes between tumor and normal tissues. B The matrix of PAM-based IE patterns with a k-value of two. C PCA shows the distribution of KIRC samples in PAM-based IE patterns. D The survival curves of KIRC samples in PAM-based IE patterns
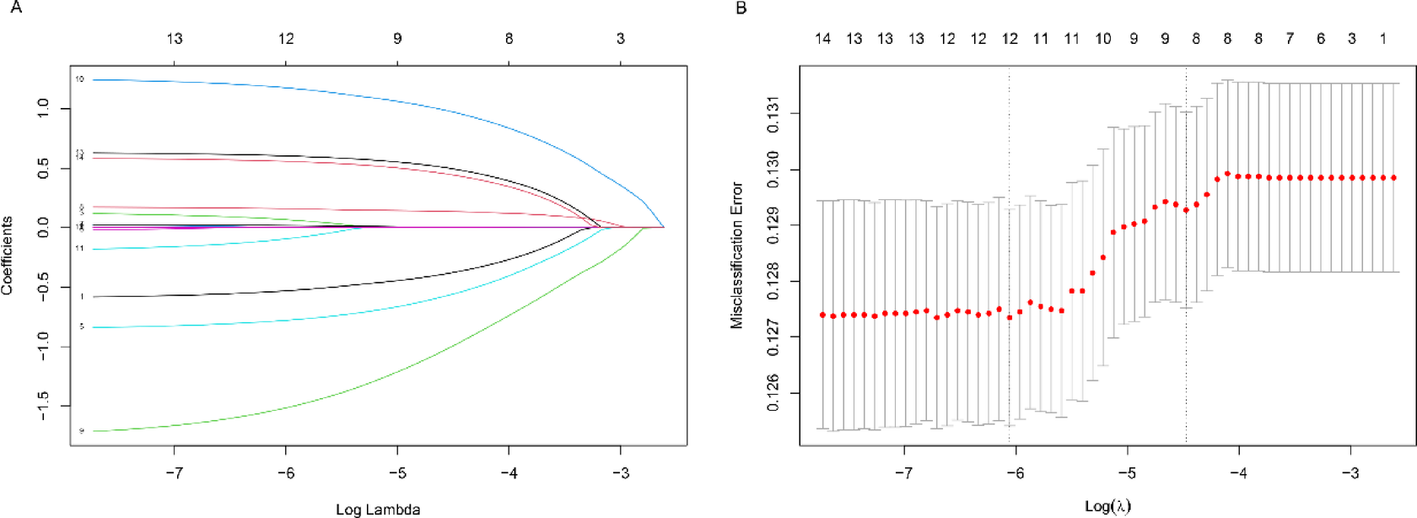
3.3 Identification of CTSS as a critical tumor suppressorThe nine intersected genes were found among malignant genes (DEGs between tumor cells and non-cancerous cells at scRNA-seq level), IE pattern-related genes (DEGs between two IE patterns), and hdWGCNA genes (green module genes) (Fig. 3A). Univariate Cox regression analysis revealed nine prognostically important genes (Fig. 3B). Three ML approaches were employed to refine the prognostic gene set from the initial nine candidates: CoxBoost regression identified six key genes (Fig. 3C), Random Survival Forest analysis selected five optimal predictors (Fig. 3D), and LASSO regression preserved seven discriminative features (Fig. 3E). These dimension reduction techniques collectively facilitated the identification of core prognostic biomarkers while maintaining model robustness. As a result, CTSS was found to be the most potent biomarker. Surprisingly, KIRC samples with high CTSS expression had significantly prolonged survival time in the TCGA-KIRC cohort (Fig. 3F), GSE29609 cohort (Fig. 3G), and E-MTAB-1980 cohort (Fig. 3H).
Fig. 3
Identification of CTSS as a critical tumor suppressor. A Venn plot shows the intersected genes among malignant genes (DEGs between tumor cells and non-cancerous cells at scRNA-seq level), pattern genes (DEGs between PAM-based IE patterns), and hdWGCNA genes (green module genes). (B) Univariate Cox regression analysis on the intersected genes. C CoxBoost shows the feature genes of prognostic intersected genes. D Random Survival Forest shows the feature genes of prognostic intersected genes. E LASSO shows the feature genes of prognostic intersected genes. F Survival curves of CTSS-based groups in the TCGA-KIRC cohort. G Survival curves of CTSS-based groups in the GSE29609 cohort. H Survival curves of CTSS-based groups in the E-MTAB-1980 cohort
3.4 Functional annotation of CTSSGO terms, including B cell activity, T cell activity, natural killer cell cytotoxicity, inflammatory activity, and immune response, were positively related to CTSS (Fig. 4A). Metascape-based pathways, including immune activity and inflammation activity, were also positively associated with CTSS (Fig. 4B).
Fig. 4
Functional annotation of CTSS. A GSEA of GO terms related to CTSS. B Metascape-based pathways related to CTSS
3.5 Mutation characteristics and drug prediction of CTSSThe mutation sites in CTSS-based groups are shown in Fig. 5A. G-score of mutation sites in CTSS-based groups revealed that 5q35.3, 5q31.3, and 5q23.2 were highly mutated in KIRC samples with high CTSS expression, while 5q35.3 and 6q21.1 were highly mutated in KIRC samples with low CTSS expression (Fig. 5B). Eight drugs, including Navitoclax, AZD1332, SCH772984, AZD4547, Ibrutinib, Acetalax, Ulixertinib, and ABT737, had significantly higher responses in KIRC samples with high CTSS expression (Fig. 5C).
Fig. 5
Mutation characteristics and drug prediction of CTSS. A Bubble plot shows the mutation sites in CTSS-based groups. B G-score of mutation sites in CTSS-based groups. C Estimated AUC of responses to eight drugs in CTSS-based groups
3.6 Immune features of CTSSCTSS is lowly expressed in tumor cells (Figure S1). Most steps of the cancer immune cycle were highly activated in KIRC samples with high CTSS expression (Fig. 6A). CTSS had significantly positive correlation with microenvironment scores (stromal, immune, estimate), TIMER-based immune cells (B cells, CD4 T cells, CD8 T cells, neutrophils, macrophages, dendritic cells), Pornpimol-based immune cells (macrophages, B cells, T cells, natural killer cells), and MCPcounter-based immune cells (T cells, B cells, neutrophils) (Fig. 6B). Besides, CTSS was significantly negatively associated with immune modulators, such as CD40, PDCD1, CTLA4, TIGIT, CD276, CD80, BTLA, HAVCR2, and CD274 (Fig. 6C).
Fig. 6
Immune features of CTSS. A Cancer immune cycles in CTSS-based groups. B The correlation between microenvironment scores, TIMER-based immune cells, Pornpimol-based immune cells, and MCPcounter-based immune cells and CTSS. C The correlation between CTSS and immune modulators
3.7 Immunotherapy prediction of CTSSThe AUC values of CTSS in predicting immunotherapy responses were larger than 0.6 in nine cohorts (Fig. 7). In addition, the levels of nine immunotherapy determinants, including CYT, IFNyIS, AyersExplS, GEP, RohIS, DavoliIS, RIR, ImmuneScore, and ICBnetIS, were all significantly higher in KIRC samples with high CTSS expression (Fig. 8).
Fig. 7
Immunotherapy prediction of CTSS. The ROC curves of CTSS’s predictive values in nine cohorts
Fig. 8
The levels of nine immunotherapy determinants in CTSS-based groups
Comments (0)