2.1 Materials
Anti-NRP-1 monoclonal antibodies (A6-11-26, designed by Dr. J. Yan) were prepared by our lab [12]. Goat anti-mouse IgG antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Three human HCC cell lines (Bel-7402, SMMC-7721, and HepG2) were provided by Cancer Research Center, Medical College of Xiamen University (Xiamen, China) after being purchased from Stem Cell Bank, Chinese Academy of Sciences (Shanghai, China). Female nude mice (aged, 6–8 weeks) were purchased from the Experimental Animal Center of Xiamen University (Xiamen, China).
2.2 Cell culture
According to routine protocols, three cell lines including Bel-7402, SMMC-7721 and HepG2 were cultured in DMEM supplemented with 10% FBS (Gibco, Thermo Fisher Scientific) at 37 °C with 5% CO2 in a humidified atmosphere.
2.3 Cell growth inhibition test
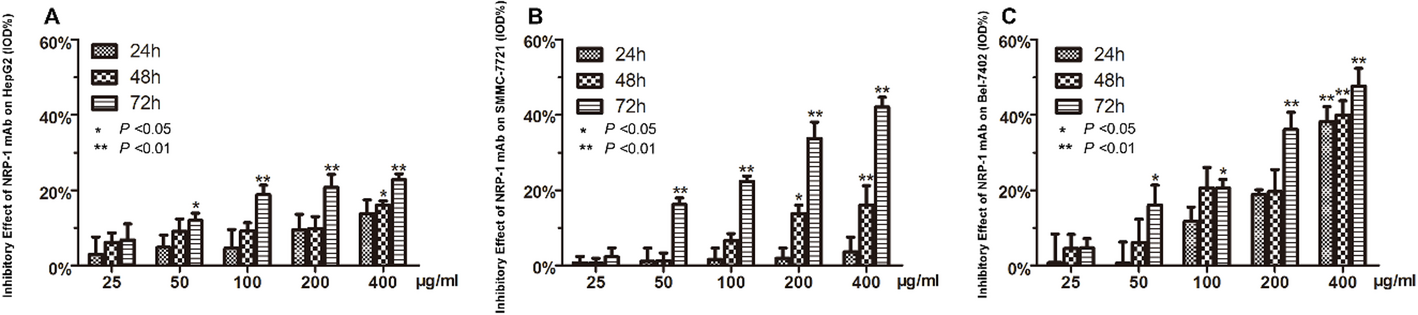
In vitro cell growth inhibition was performed by MTT test. Three HCC cell lines, Bel-7402, SMMC-7721 and HepG2 (3.0 × 104 cells/well) were seeded in 96 well plates, respectively. After 24 h, cells were incubated with various concentrations (0, 25, 50, 100, 200, 400 µg/mL) of A6-11-26 at 37 °C for 24, 48 and 72 h, respectively. Subsequently, 20 µL of 3-(4,5-dimethylthiazol-2-yl)−2,5-diphenyltetrazolium bromide (MTT, Sigma- Aldrich, St Louis, Missouri, USA) solution at a concentration of 5 mg/mL was added to each well and incubated additionally for another 4 hours. Absorbance was measured at 492 nm using a scientific microplate reader (Multiskan Spectrum; Thermo Fisher, USA). The percentage of inhibition (%) was determined using the formula:
$$\text (\%) = [(1-\text_)})/\text_)}] \times 100\%.$$
2.4 Observation of morphological changes by light microscopy
The three HCC cell lines, Bel-7402, SMMC-7721 and HepG2 (3.0 × 104 cells/well) were seeded in 96 well plates for 24 h, respectively, and then were treated with various concentrations (0, 25, 50, 100, 200, 400 µg/mL) of A6-11-26 for 24 h, 48 h and 72 h. After then, the morphological changes of these cells were observed by phase contrast microscopy (Leica, Nusslich, Germany).
2.5 Colony inhibition test
The three HCC cell lines, Bel-7402, SMMC-7721 and HepG2 (1.0 × 103 cells/well) were seeded in 6 well plates for 24 h, respectively, and then were treated with various concentrations (0, 25, 50, 100, 200, 400 µg/mL) of A6-11-26 for 2 weeks. Next, these cells were fixed by methanol and stained with 0.5% crystal violet. The number of colonies was counted manually under a microscope.
$$\begin\text &= \text) \times 100\%, \\ \text &= (1 - \text \times 100\%.\end$$
2.6 Cell apoptosis assay
Bel-7402, SMMC-7721 and HepG2 cells (5.0 × 105 cells/well) were cultured with various concentrations (0, 25, 50, 100, 200, 400 µg/mL) of A6-11-26 at 37 °C for 48 h, stained with 10 µL Propidium Iodide (PI) and 5 µL Annexin V-FITC (Becton, Dickinson and Company) and then incubated for 15 min in the dark. Cell apoptosis was analyzed by flow cytometry (Partec GmbH).
2.7 Preparation of HCC animal models and therapy
The animal study protocol has been approved by the Institutional Animal Care and Use Committee of Zhongshan Hospital Xiamen University and was conducted in accordance with the ARRIVE guidelines. The three different HCC cell lines, Bel-7402, SMMC-7721 and HepG2 (1.0 × 106 cells) were implanted subcutaneously in right lower limb of nude mice, respectively. Tumors were allowed to grow to 0.8–1.0 cm in diameter and then the tumor-bearing mice were randomly divided into 3 groups (n = 3 in each group): PBS group, low dose (1 mg/kg) group and high dose (5 mg/kg) group. A6-11-26 was given by tail vein injection every 2 d for 5 times. The body weight and tumor size were observed and recorded every 2 days. After 28 days of observation, the mice were sacrificed, and tumors were removed and weighed. The tumor volume was calculated as follows: The tumor volume = length × width × height × π/6. The growth curves of the xenograft tumors of HepG2, SMMC-7721 and Bel-7402 were documented. The tumor volume change rate (TVR%) were calculated according to the formula, TVR% = (tumor volume treated - first tumor volume)/(first tumor volume) × 100%. The tumor growth index ratio (TGI%) were calculated according to the formula:
$$\text\% = (1- \text \times 100\%.$$
2.8 Immunohistochemical analysis
The tumor tissues were harvested and immediately frozen in dry ice. After that, thawed tumor tissues were sliced into 5 μm pieces of tumor slices. After being blocked with 2% BSA (Bovine serum albumin) in phosphate-buffered saline (PBS, pH = 7.4), these slides were incubated with 1:100 diluted anti-NRP-1mAb (A6-11-26) and anti-VEGF mAb (Sigma), respectively, for 12 h at 4℃, After being washed, the slides were then incubated for 2 h at room temperature with secondary antibodies goat anti-rat IgG antibody (1:1000; Sigma). These slides were then examined and photographed using an inverted microscope (Moticam 2005, Motic). The results were analyzed using Image-Pro Plus 6.0 (Media Cybernetics, Inc.).
2.9 Western blot
The tumor tissues were sonicated and homogenized with modified Davidson Fluid (mDF) (0.5 mL) containing 1% Triton X-100 (Thermo Fisher Scientific). After centrifugation, the supernatant portions (40 µg) were subjected to SDS-PAGE gel and transferred onto a polyvinylidene fluoride membrane (Millipore, Billerica, Massachusetts, USA). To avoid interference between different antibodies, the membrane was cut to sizes that cover the internal reference and target bands respectively for transfer, followed by antibody incubation. Membranes were blocked with 2% BSA for at least 1 h, followed by overnight incubation with A6-11-26 (1:100). After extensive washing, blots were then incubated with goat anti-mouse IgG HRP secondary antibody (1: 1000; Sigma) and visualized by the ECL detection reagent (Roth, Karlsruhe, Germany) and data were analyzed with LAS 3000 digital software (Fujifilm Life Science, Santa Clara, California, USA). GAPDH blotting was used as an internal control.
2.10 Quantitative real-time PCR (qRT-PCR)
Total RNA isolated from tumor tissues was extracted with TRIZOL reagent according to the manufacturer’s instructions. The cDNA was synthesized from 1 µg of total RNA from each sample using MMLV transcriptase (ToYoBo, Shanghai, China) with random primers. Semi-Quantitative RT-PCR was performed with 2 × Phanta Max Master Mix (Vazyme, Nanjing, China). The qRT-PCR procedures were as follows: Briefly, the template cDNA was first denatured at 95 °C for 3 min. The amplification step included 25 cycles of denaturation for 15 s at 95 °C, 15 s of annealing at 60 °C, and elongation at 72 °C for 30 s.
Primers used in this study were:
NRP-1-RT-F (Homo sapiens): 5′-AAATGCGAATGGCTGATTCAG-3′;
NRP-1-RT-R (Homo sapiens): 5′-CTCCATCGAAGACTTCCACGTAGT-3′;
β-actin-RT-F (Homo sapiens): 5′-CAAGAGATGGCCACGGCTGCT-3′;
β-actin-RT-R (Homo sapiens): 5′-TCCTTCTGCATCTGTCGGCA-3′.
2.11 Statistical analysis
All the statistical analyses were performed using the SPSS 18.0 (SPSS Company, Chicago, USA). Statistical data were shown as mean ± standard deviation. For data where variables fail to meet the normality assumption based on the Shapiro-Wilk test, non-parametric tests were used for analysis. Non-parametric tests were used to compare the differences between groups of different HCC cell lines, as well as to compare the different dosages or time points within the same cell line. In vivo transplanted tumor model, non-parametric tests were used to analyze the differences in growth parameters between groups. GraphPad Prism 9.0 (GraphPad Software California, USA) was utilized to draw statistical charts. A P value < 0.05 was defined as statistically significant.

Comments (0)