
Remember me
PEComas represent a paradigm of mTOR-driven oncogenesis, with their biological complexity underscored by the interplay of molecular alterations and tumor microenvironment dynamics [15]. Our case of a metastatic TSC2/KRAS co-mutated PEComa with partial response to everolimus provides critical insights into three key areas: mechanistic foundations of mTOR dependency, biological implications of genetic heterogeneity, and therapeutic challenges in advanced disease.
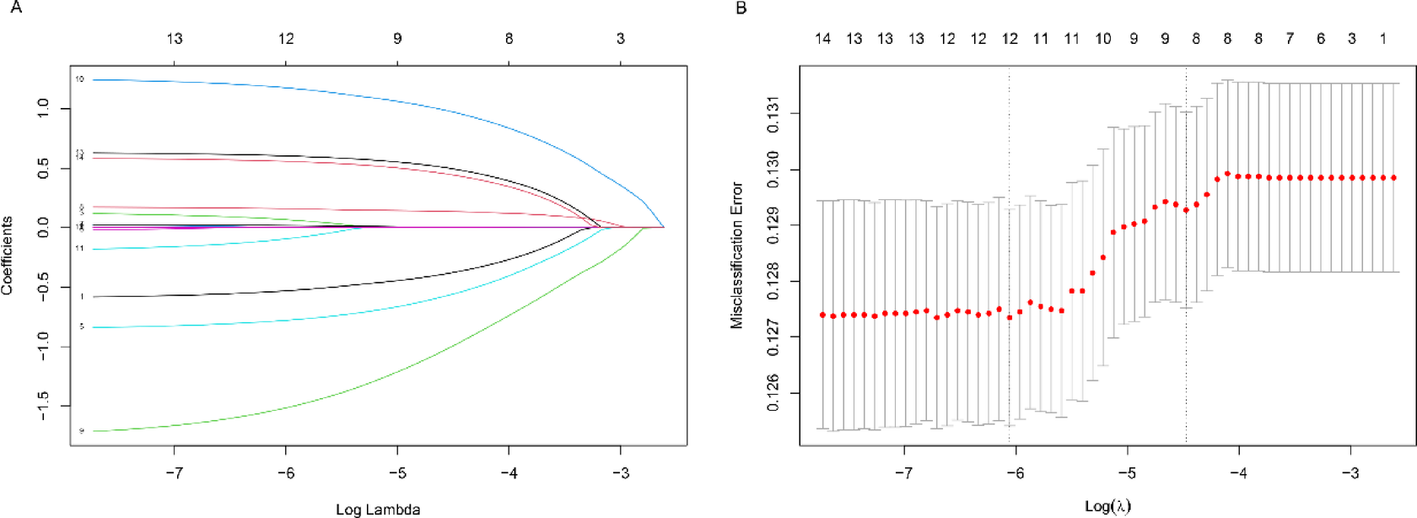
3.1 Molecular drivers and therapeutic targetingThe discovery of mechanistic link between TSC1/2 inactivation and mTOR hyperactivation has significantly advanced the management of PEComa. The TSC complex serves as a critical negative regulator of mTORC1 by hydrolyzing Rheb-GTP, and its loss results in dysregulated protein synthesis and uncontrolled cell proliferation [16, 17] (Fig. 3). In this case, the identified TSC2 mutation (68.57% VAF) is consistent with genomic studies demonstrating TSC1/2 alterations in 60–80% of malignant PEComas, which are associated with increased sensitivity to mTOR inhibitor [7]. The pivotal EXIST-2 trial—which evaluated everolimus in TSC-associated angiomyolipomas, a PEComa-family neoplasm—established the clinical rationale for mTOR inhibition, reporting significant response rates (42% vs. 0%) and prolonged progression-free survival (HR = 0.08, P < 0.0001) [18]. Expanding upon this, real-world evidence from larger PEComa cohorts reinforces mTOR inhibition efficacy. Sanfilippo et al.. documented an ORR of 41% and disease stabilization in 36% of advanced PEComa patients treated with mTOR inhibitors (sirolimus/everolimus), further validating their therapeutic role across primary site [19]. Similarly, Dickson et al.. reported sustained clinical benefit in 80% of malignant PEComa patients receiving mTOR inhibitors [20]. While nab-sirolimus is FDA-approved for malignant PEComa based on the AMPECT trial (ORR: 39%, median duration of response: 10.6 months) [11], everolimus was selected here due to: (i) Global accessibility and extensive real-world safety data in TSC-driven diseases; (ii) Institutional formulary support; (iii) Uncertainty regarding nab-sirolimus efficacy in KRAS-co-mutated tumors. However, emerging evidence suggests molecular heterogeneity may influence outcomes. Argani et al.. noted inferior responses to mTOR inhibitors in TFE3-rearranged PEComas versus conventional subtypes [21], underscoring the relevance of TFE3-negative/TSC2-mutant profile in this case.
Fig. 3
Schematic representation of the TSC1/2-mTOR signaling pathway
Our case exhibited a more pronounced tumor reduction (57.6% pulmonary, 29.1% abdominopelvic) than anticipated within 4.5 months. This aligns with reports of hyperresponsiveness in TSC2-mutant tumors, such as Testa et al.. where TSC2-inactivated PEComas showed deeper regression kinetics to mTOR inhibition than TSC1-altered counterparts [22]. This partial response may be linked to the elevated TMB (19.7 mut/Mb), as emerging evidence suggests that hypermutated tumors display increased mTOR dependency due to proteotoxic stress caused by misfolded proteins [23]. Although TMB is traditionally associated with immunotherapy response, its potential role in predicting mTOR inhibitor efficacy requires further prospective validation, particularly in light of the MSS status and low PD-L1 expression (CPS 10).
The co-occurrence of a KRAS G12D mutation (35.48% VAF) adds biological complexity to this case. While KRAS mutations are rare in PEComas, they may influence therapeutic outcomes through crosstalk between the MAPK and mTOR pathways [24]. Preclinical models indicate that KRAS-driven ERK activation can phosphorylate and destabilize TSC2, paradoxically suppressing mTORC1 activity while concurrently activating PI3K/AKT signaling [25, 26]. This dual regulatory effect may explain the sustained sensitivity to everolimus observed here, while also raising concerns about potential resistance, as KRAS-mutant tumors are known to exhibit reduced durability of mTOR-targeted therapies [27,28,29]. Importantly, longitudinal ctDNA monitoring, as recommended in recent guidelines, may enable the early detection of resistant clones and inform timely therapeutic adjustments [30].
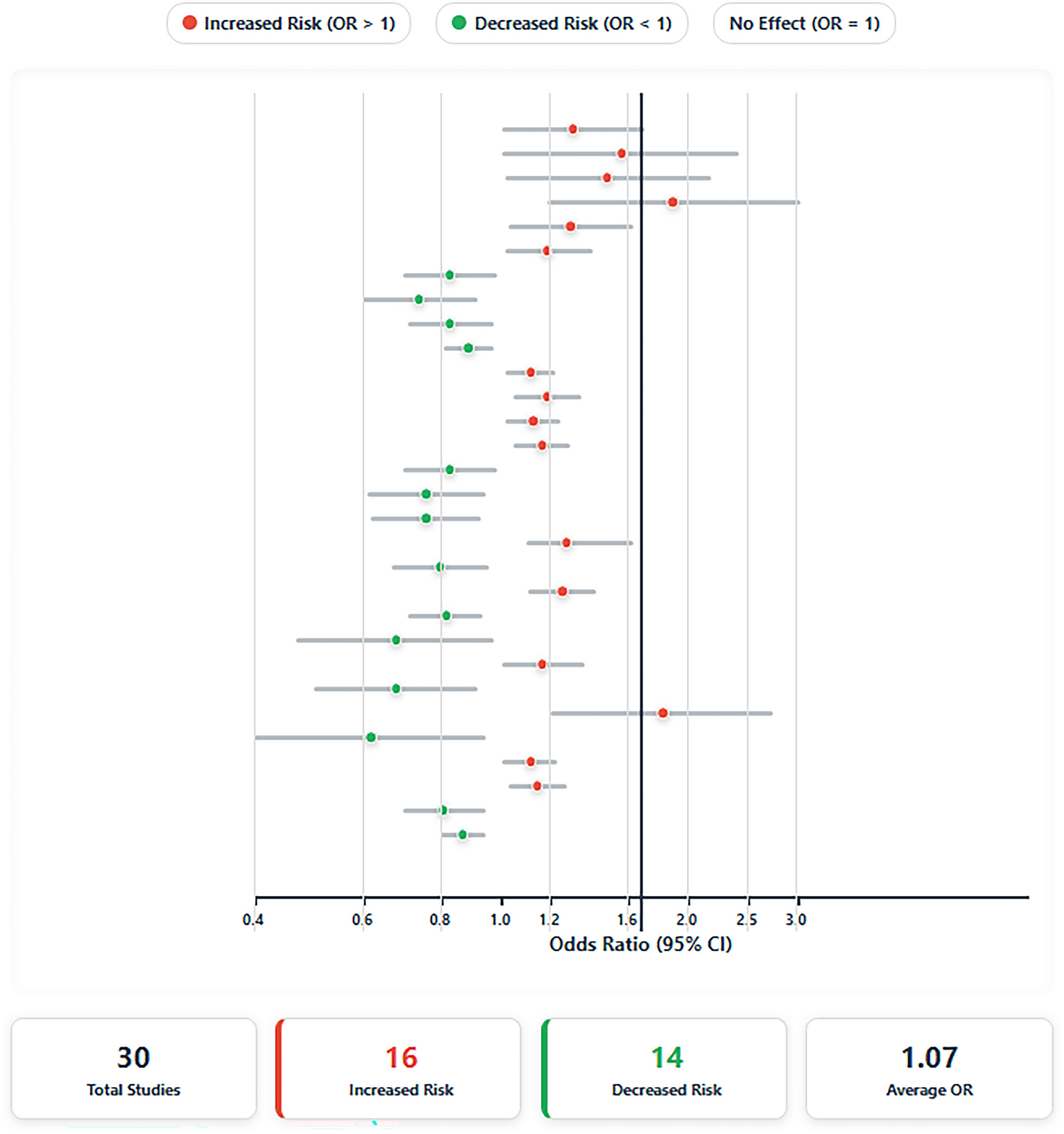
3.2 Genetic heterogeneity and clinical implicationsPEComas demonstrate substantial genetic heterogeneity extending beyond TSC1/2 alterations. Agaram et al.. reported recurrent TP53 mutations (63%) and chromatin rearrangement events involving TFE3 (23%) and RAD51B (8%) [7], which collectively underlie the spectrum of clinical behaviors observed in these tumors, ranging from indolent lesions to aggressive metastatic disease. The elevated Ki-67 proliferation index (80%) and rapid progression observed in this case align with genomic evidence linking TSC2 loss to increased mitotic activity and metastatic propensity [31]. Furthermore, the co-occurrence of KRAS and TSC2 mutations challenges conventional diagnostic frameworks. Although PEComas are histologically defined by melanocytic (HMB45/MelanA) and smooth muscle (SMA) markers, molecular profiling has become indispensable for differential diagnosis, particularly in tumors with atypical immunohistochemical features or aberrant metastatic patterns.
Notably, while immunohistochemical analysis revealed TFE3 positivity in this case, comprehensive molecular profiling did not detect a TFE3 gene fusion. This discordance underscores a critical diagnostic nuance: TFE3 immunostaining alone is not pathognomonic for TFE3-rearranged PEComa. Non-specific cytoplasmic or weakly nuclear TFE3 reactivity may occur in conventional PEComas and other mesenchymal neoplasms due to non-fusion-related mechanisms, including mTOR pathway hyperactivation or epigenetic dysregulation [15, 32]. In this context, the definitive exclusion of a TFE3 rearrangement by molecular analysis—coupled with the presence of a pathogenic TSC2 mutation, co-expression of melanocytic (HMB45/MelanA) and smooth muscle (SMA) markers, and absence of TFE3 fusion transcripts—robustly supports classification as a conventional PEComa rather than a TFE3-rearranged variant. This distinction carries clinical implications, as TFE3-rearranged tumors may exhibit distinct metastatic patterns and differential therapeutic responses.
3.3 Immunohistochemical pitfalls and diagnostic clarificationThe immunohistochemical profile in this case warrants specific discussion regarding potential diagnostic mimics. Focal CDK4 positivity in the abdominal mass and isolated MDM2 overexpression in retroperitoneal lesions could theoretically raise consideration of dedifferentiated liposarcoma (DDLPS), particularly given CDK4/MDM2 co-amplification is a hallmark of DDLPS [33]. However, three key clinicopathological and molecular features definitively refute this differential: (i) NGS revealed a pathogenic TSC2 mutation (68.57% VAF), which is a genomic signature of PEComas but absent in DDLPS. DDLPS is instead characterized by MDM2 amplification (12q14-15), which was conspicuously absent in our NGS analysis. (ii) Both primary and metastatic lesions demonstrated strong, diffuse co-expression of melanocytic markers (HMB45, MelanA) and SMA. DDLPS lacks this immunoprofile, typically showing negative melanocytic markers and variable SMA expression. (iii) Pulmonary nodules were unequivocally MDM2-negative by IHC, inconsistent with DDLPS. CDK4 expression was focal (not diffuse) and occurred without concomitant MDM2 amplification—invalidating its specificity for DDLPS. Aberrant CDK4 expression is recognized in 15–20% of sarcomas, including PEComas, often reflecting cell cycle dysregulation rather than 12q amplification [34, 35]. This integrated diagnostic approach—prioritizing TSC2 mutational status, lineage-specific IHC, and stringent interpretation of “soft tissue sarcoma” markers—robustly confirms PEComa and eliminates DDLPS from consideration.
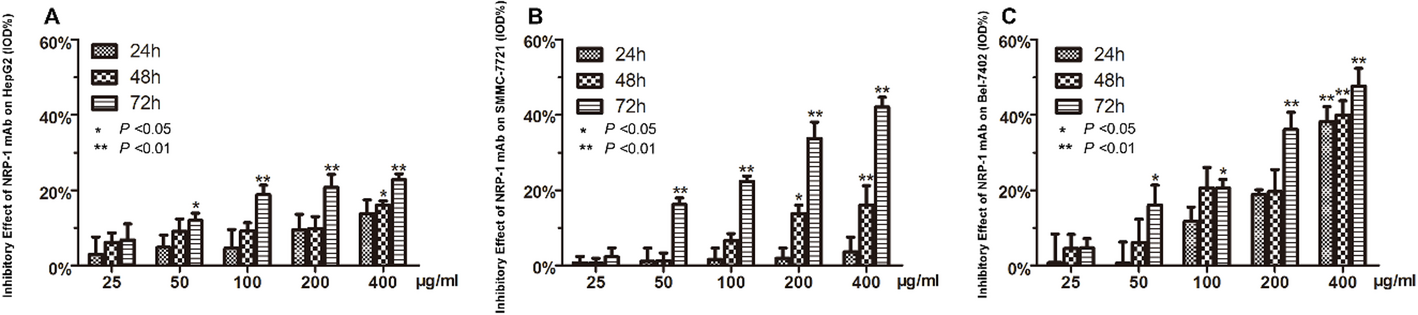
3.4 Unmet needs and future directionsDespite therapeutic advancements, PEComa management continues to present significant clinical challenges due to high recurrence rates and limited therapeutic options for refractory disease. While surgical resection remains curative for localized tumors, it is often not viable in metastatic cases such as ours. Conventional chemotherapy provides limited efficacy, with doxorubicin-based regimens demonstrating low ORR (13–20%) [19]. Targeted combination therapies represent a promising approach to circumvent resistance mechanisms: (i) Dual mTORC1/2 inhibitors (e.g., sapanisertib) have shown a 23% clinical benefit rate in everolimus-refractory advanced solid tumors, suggesting that comprehensive mTOR pathway inhibition may improve therapeutic outcomes [36]; (ii) MEK inhibitors (e.g., trametinib) exhibit preclinical efficacy in KRAS-mutant models by disrupting PD-1/PD-L1-mediated survival pathways, providing a biologically rational combinatorial strategy [37]; (iii) Autophagy inhibitors (e.g., hydroxychloroquine) synergized with everolimus in a phase I/II trial, achieving a 67% disease control rate (DCR), with 45% of DCR patients demonstrating progression-free survival exceeding 6 months [38].
This case highlights the critical need for adaptive trial designs incorporating real-time molecular monitoring. The durable response observed at 4.5 months suggests the potential for sustained clinical benefit; however, the absence of consensus guidelines for treatment duration or adjuvant therapy following initial response remains a major barrier. Emerging technologies such as patient-derived organoids (PDOs) hold substantial potential for advancing personalized therapy. Recent studies have demonstrated that PDO-based drug sensitivity testing predicts clinical responses with high predictive accuracy, enabling the optimization of tailored combination therapies for solid tumors [39, 40]. Furthermore, although the patient exhibited a favorable early treatment response, the short follow-up duration (5 months) precludes assessment of long-term outcomes. We will therefore maintain vigilant surveillance of the patient under continued everolimus therapy, including regular monitoring of tumor status, emergence of resistance, and potential serious complications. Should resistance develop, therapeutic transition to a dual mTORC1/2 inhibitor will be considered.
Comments (0)