
Remember me

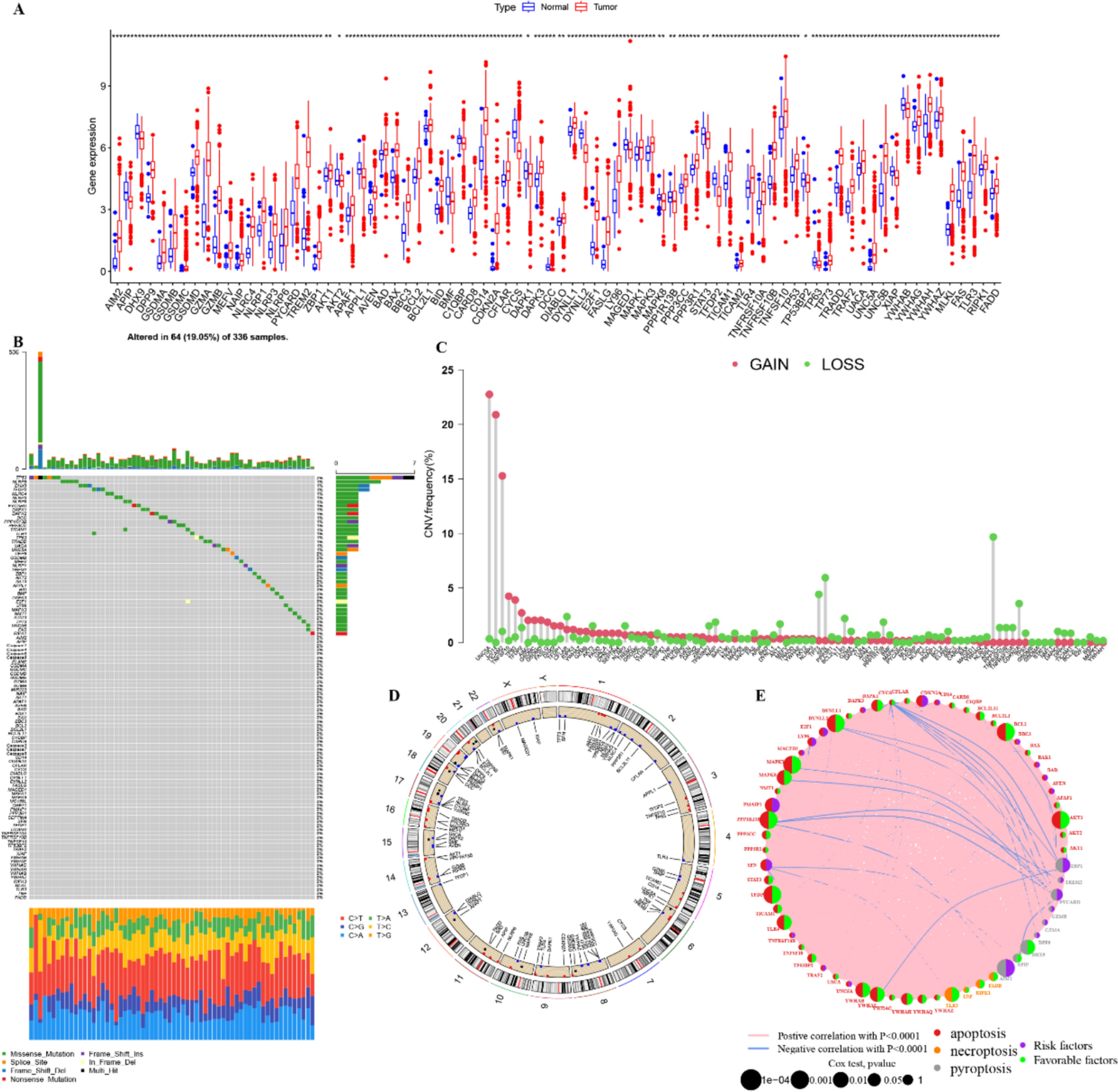
The hazard ratios of genes associated with the mTORC1 signaling pathway were calculated with 95% confidence intervals and p-values. The results indicated that EEF1E1, EIF2S2, Enolase 1(ENO1), ETF1, Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), Glutathione-disulfide reductase (GSR), GTF2H1, HSPA4, PLK1, PLOD2, Proteasome 26S subunit, non-ATPase 13 (PSMD13),, SLC1A5, SLC7A11, STIP1, TOMM40, Tubulin gamma 1 (TUBG1), ABCF2, Aurora kinase A (AURKA), BCAT1, and Cell division cycle 25A (CDC25A) were related with favorable prognosis in LIHC patients (Supplementary Figs. 1A–E).
Development of a novel prognostic-correlated survival model in LIHCInitially, the P-value threshold was set to less than 0.05, and the HR was utilized to evaluate the significance of model genes in LIHC. For instance, a POLR3G HR greater than 1 indicates that this gene acts as a risk factor for LIHC progression. Conversely, a PGM1 HR less than 1 suggests its role as a protective factor, inhibiting LIHC progression. Subsequently, these findings were further validated employing LASSO regression analysis with cross-validation, risk scores were derived using Cox regression coefficients and gene expression data. LIHC patients were divided into high-risk and low-risk groups based on the median risk score. ROC curves were constructed, and survival analyses were performed for validation. (Supplementary Figs. 1F–H). The results demonstrated that the high-risk score group was correlated with poor prognosis in LIHC patients.
OS and variation of model genes in pan-cancerTo investigate the biological functions of risk genes correlated with the mTORC1 signaling pathway across pan-cancer types, we investigated the relationship between overall survival outcomes and the expression levels of risk model-associated genes in diverse tumor cohorts from the TCGA database. A heatmap depicting the overall survival of model genes was constructed (Supplementary Fig. 2A). Elevated expression levels of these model genes were correlated with adverse clinical outcomes in LIHC patients. Additionally, TUBG1 functioned as an oncogene in THYM, THCA, PCPG, and READ. Subsequently, we explored CNV and SNV data across 33 cancer types using the TCGA database. Visualization of the variant data was performed. Notably, TPI1 predominantly exhibited heterozygous amplification in tumors such as ACC, TGCT, and OV, suggesting that TPI1 may undergo amplification in only one allele in these cancers. PLOD2 showed heterozygous amplification in CESC, UCEC, BLCA, ESCA, LUSC, and OV, while displaying heterozygous deletion in PCPG, indicating single-copy loss in these tumors. POLR3G was primarily characterized by heterozygous amplification in ACC and heterozygous deletion in BLCA, ESCA, TGCT, LUSC, and OV. Furthermore, we examined SNV of the 13 risk model genes across pan-cancer types. PLOD2 exhibited higher mutation frequencies in UCEC, SKCM, and STAD, while PLK1 showed higher mutation frequencies in UCEC and SKCM (Supplementary Figs. 2B–C).
Methylation, drug sensitivity and immune infiltration of model genes in pan-cancerWe analyzed the methylation differences of the 13 risk model genes across pan-cancer types (Supplementary Fig. 2D). The results revealed that ENO1 and PLOD2 exhibited lower methylation levels in KIRC, while PLOD2 showed higher methylation differences in LUSC. Additionally, we investigated the association between these risk model genes and critical molecular pathways implicated in tumor development. (Supplementary Fig. 2E). We found that PLK1 and POLR3G can promote apoptosis and cell cycle pathways, with POLR3G also inhibiting epithelial-mesenchymal transition (EMT), a critical biological process in tumor initiation and progression. Furthermore, to investigate the relationship between mRNA expression levels of the 13 risk model genes and drug sensitivity, we used R software to plot the association between gene expression levels and drug sensitivity (Supplementary Fig. 2F). The results demonstrated that the expression levels of PLOD2 and G6PD were positively correlated with GSK2126458 (a dual PI3K/mTOR inhibitor), while PLOD2 expression was negatively associated with the multi-target kinase inhibitor Midostaurin. Evaluating the association between risk model gene expression and anticancer drug sensitivity offers critical guidance for clinical treatment strategies and therapeutic agent selection.
Aberrant activation of molecules associated with the mTORC1 signaling pathway plays a critical role in antitumor immunity [24, 25]. To perform immune infiltration analysis, we utilized the "GSVA" package in R software, integrating it with the ssGSEA algorithm, and employed RNA-seq data in TPM format from TCGA-LIHC. A Spearman correlation analysis was conducted to evaluate the association between POLR3G expression and the infiltration levels of distinct immune cell populations. The results indicated that KIRC and HCC triggered significant infiltration of immune cells. We noticed positive correlations between tumor-infiltrating immune cells and these risk model genes, such as nTreg, Th1, B cells, and DCs in LIHC, additionally, we investigated the correlation between the risk model genes and the infiltration of major immune cells in LIHC, analyzing the associations between the 13 risk model genes and six primary tumor-infiltrating immune cell types, we found that TUBG1, SLC2A1, PLK1, LDHA, and G6PD were negatively correlated with macrophages, while POLR3G showed a positive correlation with NK cells (Supplementary Figs. 2G–H).
Protein expression levels of model genes in tissuesTo examine the protein expression profiles of the risk model genes in LIHC, we retrieved data from the HPA database for 12 genes, comparing their protein expression profiles between LIHC tissues and normal liver tissues (Supplementary Figs. 3). The results demonstrated that EEF1E1, G6PD, PLOD2, POLR3G, SLC1A5, and TUBG1 exhibited higher expression levels in HCC compared to liver tissues. Further in vivo and in vitro studies, along with mechanistic exploration of these model genes in LIHC, may reveal their potential as critical therapeutic targets for future LIHC treatment strategies.
Elevated expression levels of POLR3G are correlated with poor prognosis in patients with LIHCThrough comparative analysis of POLR3G expression between unpaired and paired LIHC tumor samples and adjacent normal tissues. The findings demonstrated a marked upregulation of POLR3G expression in tumor tissues (Fig. 1A, 1B). ROC curve analysis demonstrated that POLR3G is effective for the diagnosis of LIHC (Fig. 1C). Subsequent survival analysis indicated that elevated POLR3G expression profiles are correlated with poor prognosis in LIHC patients (Fig. 1D–F).
Fig. 1

A, B Differences in the expression of POLR3G across pan-cancer. Transcriptional profiling and clinical significance of POLR3G in hepatocellular carcinoma. C, D Comparative analysis of POLR3G expression patterns between tumor specimens and adjacent normal tissues in TCGA-LIHC cohort, including both paired and unpaired samples. E Receiver operating characteristic (ROC) analysis demonstrating the diagnostic efficacy of POLR3G in discriminating malignant from non-neoplastic hepatic tissues. F–H The survival curve showed that the higher the expression level of POLR3G in LIHC patients, the worse the DSS, PFI and OS of the patients. (*p < 0.05, **p < 0.01, ***p < 0.001)
POLR3G is associated with the clinical characteristics of LIHC patientsWe examined the association between POLR3G expression and clinicopathological features (Fig. 2A–I). POLR3G expression levels were significantly associated with tumor grade, patient age and gender, OS events, DSS events, and PFI events. Higher tumor grades correlated with increased POLR3G expression levels.
Fig. 2
The correlation between POLR3G expression level and clinicopathological features in LIHC patients. A Pathologic N stage, B Pathologic M stage, C Pathologic stage, D Pathologic T stage, E Gender, F Age, G DSS event, H PFI event, I OS event. (*p < 0.05, **p < 0.01, ***p < 0.001)
Pathway and functional enrichment analysis of POLR3G-associated differential genesWe investigated genes co-expressed with POLR3G in LIHC, identifying those positively or negatively correlated with its expression. The heatmap displays the top 10 positively and negatively correlated genes, along with their expression levels in LIHC (Fig. 3A–D).
Fig. 3
A-D Heatmap of the correlation between the top ten related genes to positive and negative in high and low POLR3G expression groups. (*p < 0.05, **p < 0.01, ***p < 0.001)
Using GeneMANIA, we analyzed 20 genes associated with POLR3G (Fig. 7A). The STRING database was employed to predict the PPI network related to POLR3G (Fig. 4B). GO and KEGG enrichment analyses of differentially expressed genes (DEGs) demonstrated that enriched biological processes (BP) included transcription by RNA polymerase III, DNA-templated transcription initiation, ncRNA transcription, rRNA transcription, and rRNA metabolic process (Fig. 4C). Enriched cellular components (CC) comprised the DNA-directed RNA polymerase complex, nuclear DNA-directed RNA polymerase complex, RNA polymerase complex, transferase complex transferring phosphorus-containing groups, and RNA polymerase III complex (Fig. 4D). Molecular functions (MF) were primarily associated with DNA-directed 5’-3’ RNA polymerase activity, RNA polymerase activity, 5’-3’ RNA polymerase activity, nucleotidy transferase activity, catalytic activity, and activity acting on RNA (Fig. 4E). KEGG pathway enrichment predominantly involved RNA polymerase, the cytosolic DNA-sensing pathway, Huntington’s disease, basal transcription factors, and viral carcinogenesis (Fig. 4F).
Fig. 4
Correlation and enrichment analysis of POLR3G-related genes and proteins. A PPI network showing the top twenty proteins interacting with POLR3G. B The top 100 proteins with the highest probability of interaction were selected through the STRING database. C-F GO and KEGG analysis
Possible signaling pathways involved by POLR3G in LIHCGSEA was conducted to identify the signaling pathways correlated with POLR3G-related DEGs (Fig. 5A–I). The figure summarizes the most representative enriched pathways. These results suggest that POLR3G may contribute to hepatocellular carcinoma progression through pathways such as Fatty Acid Metabolism, Respiratory Electron Transport, Mitochondrial Complex IV, Electron Transport Chain Oxphos System in Mitochondria, Sphingolipid De Novo Biosynthesis, Galanin Receptor Pathway, Oxidative Phosphorylation, and Lipid Particles Composition. This supports its possible as a tumor biomarker and therapeutic target.
Fig. 5
GSEA analysis for POLR3G in LIHC A Fatty Acid Metabolism. B Electron Transport Chain Oxphos System in Mitochondria. C Oxidative Phosphorylation. D Mitochondrial Complex IV Assembly. E Respiratory Electron Transport. F Sphingolipid De Novo Biosynthesis. G Galanin Receptor Pathway. H Lipid Particles Composition. I Role of Phospholipids in Phagocytosis
Relationship between POLR3G and immune infiltrationTo perform immune infiltration analysis, we utilized the "GSVA" package in R software, integrating it with the ssGSEA algorithm, and employed RNA-seq data in TPM format from TCGA-LIHC. Spearman correlation analysis was applied to evaluate the associated between POLR3G and the infiltration levels immune cell infiltration levels. The results demonstrated that POLR3G exhibited positive correlations with Th2 cells, Tcm, T helper cells, Adc, Macrophages, and Tgd, while showing negative correlations with Treg, Neutrophils, Tem, IDC, Th1 cells, T cells, DC, Cytotoxic cells, NK CD56bright cells, Mast cells, NK CD56dim cells, TFH, B cells, Th17 cells, Eosinophils, CD8 T cells, NK cells, and pDC (Fig. 6A). Additionally, we assessed the infiltration levels of key immune cell subsets exhibiting the strongest correlation in POLR3G high- and low-expression cohorts. The infiltration levels of NK cells, T helper cells, pDC, Tcm, Th2, and CD8 T cells showed statistically significant differences between the POLR3G high-expression and low-expression groups (Fig. 6B–G).
Fig. 6
The relationship between the expression level of POLR3G and immune cell infiltration in LIHC. A The relationship between POLR3G expression levels and immune cell abundance. B—G Comparative analysis of tumor microenvironment immunoprofiles stratified by POLR3G transcriptional status, evaluating differential immune cell infiltration patterns between high- and low-expression cohorts. (*p < 0.05, **p < 0.01, ***p < 0.001)
Knockdown of POLR3G inhibited the proliferation of LIHC cell linesTo investigate the impact of POLR3G on the proliferative ability of LIHC cells, we conducted CCK-8 and EDU cell proliferation assays (Fig. 7A–G). Following POLR3G knockdown, the proliferative ability of Huh7 and SMMC-7721 cells was significantly inhibited.
Fig. 7
Functional characterization of POLR3G knockdown on hepatocellular carcinoma cell proliferation kinetics. A-C The knockdown efficiency of POLR3G in hepatocellular carcinoma cells was validated through PCR and Western blot analyses. D-E EDU assay demonstrating proliferative ability in POLR3G-silenced Huh7 and SMMC-7721 cell lines. F-G Quantitative assessment of cell proliferation ability through CCK-8 assay following POLR3G -silenced in both hepatocellular carcinoma cells. (*p < 0.05, **p < 0.01, ***p < 0.001)
Comments (0)