
Remember me

Cancer remains one of the most significant global health challenges, contributing to high rates of morbidity and mortality. It ranks among the top three leading causes of death in individuals aged 30 to 69 years in 177 out of 183 countries [1]. According to the 2022 Global Cancer Statistics, nearly 20 million new cancer cases and approximately 10 million cancer-related deaths occurred worldwide in that year [2]. Cancer encompasses a diverse group of diseases characterized by uncontrolled and abnormal cell proliferation. These malignant cells can invade surrounding tissues and metastasize to distant organs via the blood and lymphatic systems. Metastasis is the predominant cause of cancer-related deaths [3,4,5]. Normal cellular development and tissue homeostasis rely on tightly regulated processes, including cell proliferation, differentiation, and apoptosis. Disruption in any of these processes can result in unchecked cell growth, contributing to cancer initiation and progression [6]. Cell growth is regulated through the coordinated actions of specific genes within each cell. Genomic instability plays a central role in the development of neoplastic diseases by promoting mutations and chromosomal alterations [7].
Two major categories of genes are implicated in cancer: tumor suppressor genes and oncogenes. A disruption or imbalance between these gene classes can drive abnormal cell proliferation. Oncogenes (OGCs) arise from mutations in proto-oncogenes—genes that normally regulate cell division and growth. When mutated, proto-oncogenes become oncogenes, leading to excessive protein production or increased activity beyond normal regulatory limits. This dysregulation enables cells to proliferate autonomously, disregarding the organism’s growth control mechanisms.
Tumor suppressor genes (TSGs) are essential for maintaining normal cellular homeostasis by regulating cell division and promoting apoptosis. Under physiological conditions, TSGs prevent excessive cell proliferation and ensure the removal of damaged or abnormal cells. However, when TSGs become inactivated or dysfunctional, cells can proliferate uncontrollably, bypassing the body’s regulatory mechanisms. The simultaneous activation of oncogenes (OGCs) and inactivation of TSGs create a permissive environment for unchecked cell growth and resistance to apoptosis—two fundamental hallmarks of cancer initiation and progression [8,9,10].
Advancing the identification, functional characterization, and mechanistic understanding of these gene classes is critical for progress in cancer research and clinical management. This knowledge underpins the discovery of novel biomarkers and drives the development of targeted therapies, ultimately enabling improved prevention, early detection, and personalized treatment strategies.
The WT1 gene (Wilms Tumor 1) was first identified in studies of Wilms tumor, a pediatric renal malignancy. In 1990, researchers mapped the WT1 gene to chromosome 11p13 by analyzing chromosomal abnormalities in affected patients. These findings led to the recognition of WT1 as a tumor suppressor gene, based on its frequent inactivation in Wilms tumor cases [11].
Wilms’ tumor is the most common pediatric renal malignancy, accounting for approximately 7% of all childhood cancers [12]. Genetic analyses of affected patients frequently reveal mutations in several genes, including the homozygous inactivation of the WT1 tumor suppressor gene [13, 14].
Despite being initially identified and classified as a tumor suppressor, accumulating research has demonstrated that WT1 can also function as an oncogene under certain cellular contexts, earning it the designation of a “chameleon gene.” [15]. The significance of WT1 extends beyond oncology; it plays critical roles in developmental biology, cancer progression, and clinical diagnostics. Recent studies, such as that by Jing et al. (2022), have shown that WT1 can inhibit the proliferation of human renal carcinoma cells and induce G2/M cell cycle arrest by upregulating IL-24 expression [16].
Moreover, WT1 is implicated in the pathogenesis of a broad spectrum of malignancies, including hematologic cancers and various solid tumors. These include leukemia, breast cancer, ovarian cancer, renal cell carcinoma, mesothelioma, melanoma, glioblastoma, and soft tissue sarcomas, among others [17,18,19,20,21,22,23,24].
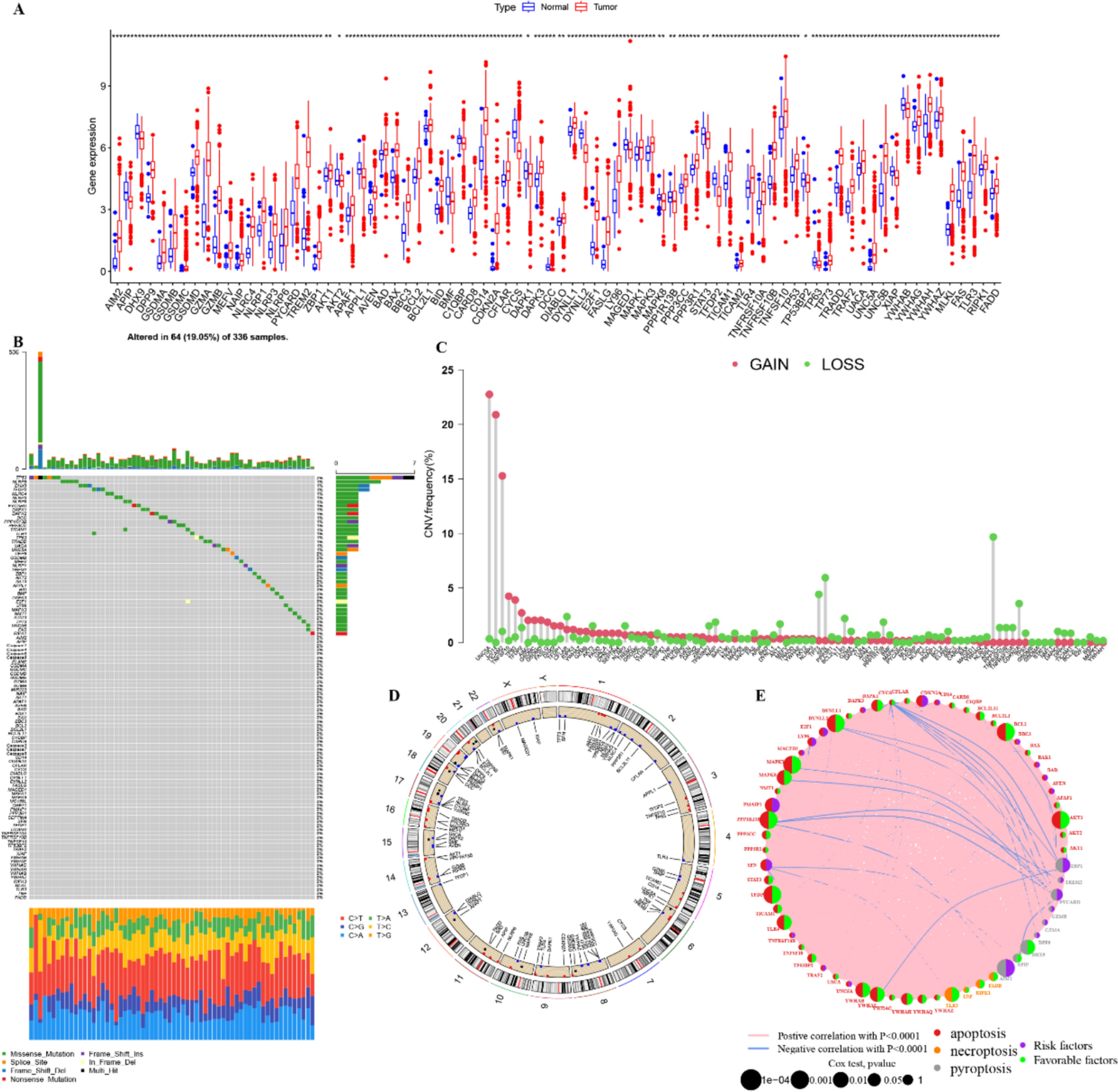
Structure of WT1 geneThe human WT1 gene, located on chromosome 11p13, is a complex locus spanning approximately 50-kb and composed of 10 exons. It encodes a 3-kb mRNA transcript [25, 26].
The WT1 gene encodes a transcription factor characterized by four zinc finger motifs at its C-terminal region, each comprising two cysteine and two histidine residues (Fig. 1). These zinc finger domains enable DNA and RNA binding, direct nuclear localization, mediate protein–protein interactions and regulate gene transcription. In contrast, the N-terminal region is enriched in proline, glutamic acid, serine, and glycine residues. This region enables WT1 to self-associate and plays a crucial role in regulating both transcriptional activation and repression [27,28,29].The WT1 gene generates multiple transcript variants through alternative splicing at two coding exons. One splicing event involves the inclusion or exclusion of exon 5, which encodes a 17-amino acid peptide, located just N-terminal to the four zinc finger motifs. Another splicing event occurs at exon 9, resulting in the inclusion or exclusion of a three amino acid sequence lysine-threonine-serine (KTS)-positioned between the third and fourth zinc fingers [30,31,32,33]. Alternative splicing produces several WT1 isoforms, with four major variants designated A, B, C, and D. Isoform A lacks both the 17–amino acid insert and the KTS sequence. Isoform B contains the 17–amino acid insert but lacks the KTS sequence. Isoform C includes the KTS sequence but does not have the 17–amino acid insertion, while isoform D harbors both the 17–amino acid insert and the KTS inserts. These isoforms exhibit distinct functional properties and regulate specific target genes, with expression patterns that are consistent yet tissue-dependent [32, 34, 35].
Fig. 1
Schematic Representation of the Structural Organization of the WT1 Protein. The C-terminus of the WT1 protein contains four zinc finger motifs, which are implicated in DNA and RNA binding. The N-terminus features a transregulatory domain. Brown circles represent the repression domain, while blue circles indicate the activation domain
Molecular function of WT1 geneWT1 regulates a broad spectrum of target genes involved in key cellular pathways such as growth signaling, cell cycle progression, differentiation, and apoptosis. By modulating these genes, WT1 plays critical roles in physiological processes including cell proliferation, survival, programmed cell death, and the maintenance of mesenchymal–epithelial balance. Its transcriptional activity is context-dependent; in some cases, WT1 functions as an activator, while in others, it acts as a repressor. This dual role highlights WT1’s versatility in regulating gene expression and cellular processes [36,37,38,39,40,41,42]. For instance, WT1 activates Wnt4 gene expression in the kidney to promote MET, while repressing its expression in the heart to regulate EMT. This context-dependent regulation is achieved through WT1’s recruitment of different cofactors: coactivators like CBP/p300 in the kidney and corepressors like BASP1 in the heart. [43]. Multiple studies have shown that WT1’s dual role as both an activator and repressor is influenced by the diversity of its isoforms, its interactions with different binding partners, and the specific cellular context in which it functions [27, 28, 44]. This functional versatility arises from the WT1 N-terminal region, which contains both a transcriptional activation domain and a suppression domain (SD), allowing it to modulate gene expression in a context-dependent manner. Importantly, a defined region within the N-terminus can inhibit WT1’s own activation domain, further contributing to its regulatory complexity [45, 46]. The suppression domain of WT1 represses the activity of other transcriptional activators by recruiting the cosuppressor, brain acid-soluble protein 1 (BASP1). BASP1 serves as a crucial component of the WT1 cosuppressor complex and plays a key role in modulating WT1’s transcriptional activity [47, 48].
The WT1 gene and its isoforms regulate genes critical for cellular differentiation and survival in multiple organs, including the spleen, adrenal glands, liver, diaphragm, gonads, kidneys, and urogenital system [49,50,51]. WT1 is also essential for heart development and regulates gene expression in cardiomyocytes to maintain cardiac homeostasis and promote repair after injury, highlighting WT1 as a promising target for cardiac regeneration therapies [52]. Quenneville et al. (2024) identified a novel WT1 isoform induced under prolonged severe hypoxia in cancer cells, which drives a non-canonical epithelial–mesenchymal transition, illustrating WT1’s functional versatility through isoform-specific gene regulation in stress conditions [53]. Additionally, studies have demonstrated that loss of WT1 function impairs the regulation of key genes required for kidney development and promotes tumorigenesis, contributing to the onset of Wilms’ tumor. These observations emphasize WT1’s critical role in regulating cellular proliferation and differentiation [54].
WT1 also contributes significantly to RNA processing and metabolism by interacting with splicing factors. In addition, it influences mRNA transport and stability. [32, 44, 55]. WT1 participates in translation process, as the WT1 protein shuttles between the nucleus and cytoplasm [56, 57]. Moreover, it regulates mRNA turnover through interactions with the 3′ untranslated region (UTR), implicating microRNA processing pathway genes in the pathogenesis of Wilms’ tumor [58].
WT1 exhibits a dual role in cancer pathogenesisIn Wilms’ tumor, WT1 primarily functions as a tumor suppressor by regulating genes essential for cellular differentiation and apoptosis. It suppresses oncogenic signaling pathways while promoting differentiation and programmed cell death in tumor cells [59,60,61]. However, the role of WT1 in other cancers is far more complex and context-dependent, influenced by the cellular environment and its interaction with key pathways such as p53. For instance, Yao et al. (2021) demonstrated that WT1 suppresses acute myeloid leukemia cell proliferation in a p53-dependent manner, highlighting its tumor-suppressive function when p53 is active [62]. Additionally, Jing et al. (2022) reported that WT1 inhibits the proliferation of renal carcinoma cells and induces G2/M arrest by upregulating pro-apoptotic genes IL-24, reinforcing its tumor-suppressive activity in renal cell carcinoma (RCC) [16].
WT1 exhibits a dual role in tumorigenesis, acting as both a tumor suppressor and an oncogene. This functional dichotomy is primarily driven by specific WT1 isoforms, which exert distinct and often opposing effects on key biological processes. Isoform expression levels and activity critically influence tumor progression and clinical outcomes, emphasizing the need for isoform-specific regulation in cancer biology [59, 63]. WT1’s oncogenic or tumor-suppressive function also depends on factors such as developmental stage, cell type, differentiation status, and the timing of its expression or mutation. Additionally, its activity is modulated by binding partners, co-occurring genetic mutations, and features of the tumor microenvironment [15, 25, 27, 28, 44].
WT1 predominantly acts as an oncogene in many adult cancers by modulating apoptotic pathway in a complex, context-specific manner. It promotes cell survival by upregulating anti-apoptotic genes and suppressing pro-apoptotic pathways. This oncogenic activity is particularly evident in leukemic cells, where WT1 fosters an anti-apoptotic environment by enhancing the expression of survival genes and repressing apoptosis, thereby contributing to its oncogenic role [64,65,66,67].Furthermore, mutations in the WT1 gene or epigenetic modifications, such as promoter methylation, can alter its transcriptional activity and shifting its role from tumor suppressive to oncogenic. The epigenetic landscape of the tumor microenvironment further modulates WT1 expression and activity, reinforcing its dual and context-dependent role in cancer pathogenesis [59, 63].
Table 1 presents the context-dependent cellular and tissue-specific functions of the WT1 gene. Originally identified as a tumor suppressor, WT1 also functions as an oncogene in various malignancies. In cancers such as acute myeloid leukemia (AML), myelodysplastic syndromes (MDS), non-small-cell lung cancer (NSCLC), and glioblastoma, WT1 promotes cell proliferation and survival by upregulating oncogenic targets, including BCL-2, MYC, VEGF, and EGFR [65, 66, 68, 69]. In these contexts, WT1 serves as a therapeutic target, particularly through immunotherapeutic strategies such as WT1 peptide vaccines. Conversely, in cancers like Wilms’ tumor, mesothelioma, and breast cancer, WT1 acts as a tumor suppressor. Loss-of-function mutations in WT1 in these cases are associated with impaired differentiation, reduced apoptosis, and deregulated cell cycle control. Under normal conditions, WT1 supports differentiation, apoptosis, and cell cycle arrest by activating targets like p21, p53, and IGF-binding proteins [15, 70]. These dual and often contradictory roles of WT1 in cancer underscore the importance of context-specific analysis in both diagnostic assessment and therapeutic development.
Table 1 WT1 as Oncogene vs Tumor SuppressorWT1 interactions with other tumor suppressorsWT1 engages in complex and context-dependent interactions with other tumor suppressor genes, thereby regulating critical cellular processes such as apoptosis, cell cycle progression, and genomic stability. Recent studies demonstrate that WT1 directly interacts with members of the p53 protein family including p53, p63, and p73—modulating their transcriptional activities and influencing cellular outcomes related to apoptosis, cell cycle control, and tumorigenesis (Tables 2 and 3).
Table 2 WT1 interactions with other cancer genesTable 3 WT1 Gene Involvement In Various Type CancersP53:WT1 physically interacts with p53 through its zinc finger domains, particularly zinc fingers 1 and 2 [71]. This interaction stabilizes p53 by extending its half-life and shielding it from proteasomal degradation. As a consequence, WT1 enhances p53’s transcriptional activity, promoting the expression of genes involved in cell cycle arrest and apoptosis [42, 44]. Despite this activation, WT1 paradoxically inhibits p53-induced apoptosis, highlighting a context-dependent regulatory complexity in their interaction [44,
Comments (0)