
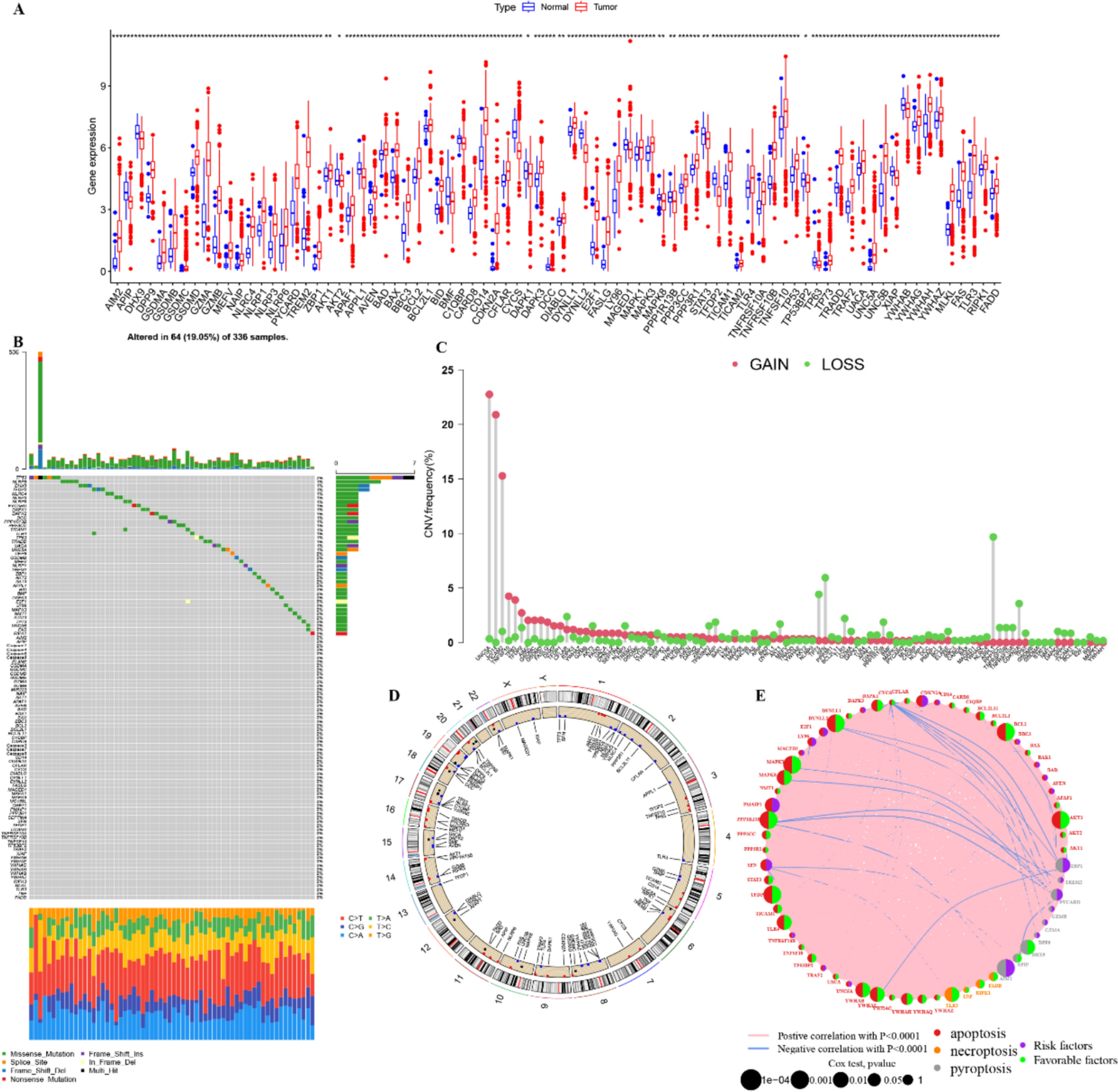
Remember me

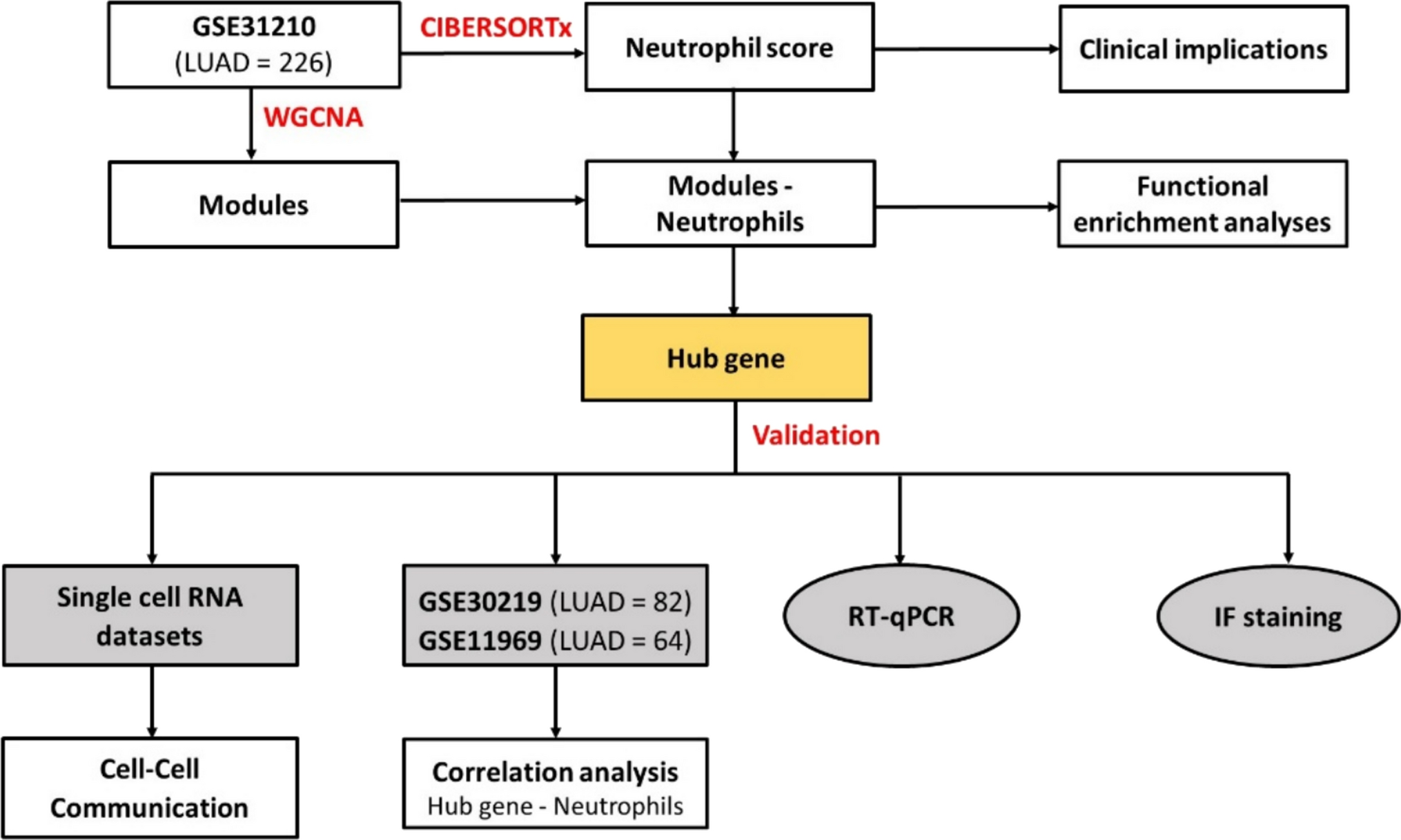
A comprehensive workflow of the analytical process is summarized (Fig. 1). The schematic outlines the full data processing pipeline, including data acquisition from GEO, DEG identification, WGCNA, machine learning-based biomarker screening, enrichment analyses, and immune cell deconvolution. It illustrates how the GSE145974 dataset was primarily used for discovery analyses, while GSE63085 served as a validation cohort. The integration of multiple analytical strategies enhances the robustness and interpretability of the study.
Fig. 1
Schematic of the study workflow. ROC: Receiver Operating Characteristic; GSEA: Gene Set Enrichment Analysis; GO: Genetic Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; AUC: Area Under the Curve; and FDR: False Discovery Rate
Weighted gene coexpression network analysisTo investigate transcriptomic modules associated with Lyme disease, we conducted WGCNA on the top 5000 most variable genes from the GSE145974 dataset. Three major modules showed significant correlations with disease status: The blue module was positively associated (r = 0.56, p = 1e–4), while the red and black modules showed negative correlations (r = − 0.57 and − 0.56, respectively) (Fig. 2a). These findings highlight discrete gene coexpression patterns linked to LD pathology. The soft-thresholding power used to ensure scale-free topology was selected as 19 (Fig. 2b).
Fig. 2
Construction of weighted gene coexpression network analysis (WGCNA) module. a Associations between gene modules and LD. Each row corresponds to a module eigengene, and each column corresponds to a clinical status. Each cell contains the correlations and p values between each module and clinical status, b the cluster dendrogram of the top 25% of genes with the highest variance in the GSE63085 dataset. Each color represents a specific gene module, wherein genes within the same module share significant biological functions, c, d, and e gene Ontology (GO)/Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses of the significant modules identified via WGCNA. WGCNA: Weighted Gene Coexpression Network Analysis and LD: Lyme disease
We then performed functional enrichment analysis of each key module. The black module was significantly enriched in GO terms related to “ubiquitin protein ligase binding,” indicating altered proteostasis mechanisms (Fig. 2c). The red module was associated with mitochondrial dysfunction and neurodegeneration pathways, such as “amyotrophic lateral sclerosis” and “respiratory chain” components (Fig. 2d). The blue module, most positively associated with LD, was enriched in processes related to neutrophil activation and degranulation, underscoring a strong innate immune activation signature (Fig. 2e).
Identification of differentially expressed genes and hub genes through machine learning algorithmsTo identify specific transcriptional differences between LD and healthy controls, we analyzed DEGs in the GSE145974 dataset. A total of 91 DEGs were identified, with 65 upregulated and 26 downregulated in LD samples (adjusted p < 0.05; |logFC|≥ 1). A heatmap of the top DEGs demonstrates clear separation between LD and control profiles (Fig. 3a). A volcano plot illustrates the global differential expression landscape, with significant genes highlighted (Fig. 3b). The upregulation of immune-related and inflammatory genes suggests that LD is associated with a pronounced pro-inflammatory transcriptional profile. These DEGs may reflect the host immune response to Borrelia infection, particularly the activation of innate immune signaling and stress response pathways. This differential expression signature provides a foundational dataset for the subsequent identification of robust biomarkers.
Fig. 3
a Heatmaps illustrating the fold change between patients with LD and controls, b a volcano plot representing differentially expressed genes. Candidate biomarkers were identified using machine learning algorithms, c and d The LASSO regression algorithm identified 15 genes as biomarkers with minimal binomial deviation, e the top 10 genes were selected and ranked based on the importance scores obtained using the random forest algorithm, f and g the support vector machine recursive feature elimination (SVM–RFE) algorithm was used for screening candidate genes. The highlighted point indicates minimal error rate and optimal accuracy, with the corresponding genes representing the best signature selected by SVM–RFE, h three hub genes (FCGR1B, MPP1, and HSPA6) were selected. A Venn diagram was used to visualize the intersection between the three machine learning algorithms. Lasso: least absolute shrinkage and selection operator and SVM-RFE: support vector machine recursive feature elimination
To identify core diagnostic markers, we applied three machine learning approaches: LASSO regression, random forest (RF), and SVM–RFE. LASSO selected 15 genes with nonzero coefficients, including FCGR1B and MPP1 (Fig. 3c–d). RF ranked the top genes by importance score, with FCGR1B and MPP1 again ranking highly (Fig. 3e). SVM–RFE identified a 36-gene optimal set (Fig. 3F–G). Intersection analysis using a Venn diagram revealed three overlapping hub genes: FCGR1B, MPP1, and HSPA6 (Fig. 3h).
The consistent identification of these three genes across independent algorithms suggests that they are not only statistically robust but also biologically meaningful. FCGR1B is known to be involved in immunoglobulin binding and monocyte signaling, MPP1 plays a role in membrane–cytoskeletal interactions and immune cell polarity, and HSPA6 is part of the heat shock protein family, implicated in cellular stress responses. Their emergence as central nodes may reflect their involvement in the host’s immune activation and cellular response to bacterial infection in Lyme disease.
Validation of diagnostic performanceTo validate the diagnostic utility of the hub genes, we evaluated their performance using ROC curve analysis. In the GSE145974 dataset, all three genes achieved strong classification performance with AUC values exceeding 0.90, indicating high sensitivity and specificity (Fig. 4a). External validation using the GSE63085 dataset confirmed robust diagnostic performance for FCGR1B and MPP1, with AUC values above 0.80 (Fig. 4b). HSPA6 could not be assessed due to missing expression data in GSE63085.
Fig. 4
Visualization of gene expression and external data verification of biomarkers. a ROC curve analysis of three hub genes in the GSE145974 dataset, b ROC curve analysis of two hub genes in the GSE63085 dataset, c box plot showing the expression levels of FCGR1B, MPP1, and HSPA6. Significance levels are indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001
In both datasets, box plot analysis demonstrated significantly elevated expression of the three hub genes in LD samples compared to healthy controls (Fig. 4c). These consistent results across independent datasets support the robustness and generalizability of the selected biomarkers.
Gene set enrichment analysis (GSEA)To gain insight into the biological roles of the hub genes, we performed single-gene GSEA for FCGR1B, HSPA6, and MPP1. Only the top three significantly enriched pathways per gene were reported and categorized into overarching biological themes (e.g., immune signaling, metabolism, and circadian regulation) to facilitate interpretation. FCGR1B was associated with upregulation of pathways such as “Proteasome,” “Collecting Duct Acid Secretion,” and “One Carbon Pool by Folate” (Fig. 5a). These pathways are involved in protein degradation, renal ion transport, and folate metabolism, suggesting that FCGR1B may play a role in metabolic–immune crosstalk. HSPA6 was enriched in neutrophil extracellular trap formation and 2-oxocarboxylic acid metabolism while being negatively associated with circadian rhythm regulation and taste transduction (Fig. 5b). MPP1 was positively associated with antifolate resistance and glycan degradation, and negatively associated with circadian signaling and hedgehog pathways (Fig. 5c). These findings imply that circadian dysregulation and altered metabolic responses are potential features of the LD transcriptome. Global GSEA of all DEGs revealed that immune-related and metabolic processes, including “proteasome,” “aldosterone-regulated sodium reabsorption,” and “circadian rhythm,” were significantly enriched, supporting the notion of systemic metabolic and inflammatory activation in LD (Fig. 5d).
Fig. 5
Functional enrichment analysis. a The main upregulated and downregulated signaling pathways significantly enriched in FCGR1B, b the main upregulated and downregulated signaling pathways significantly enriched in HSPA6, c the main upregulated and downregulated signaling pathways significantly enriched in MPP1, d the key signaling pathways of DEGs. Abbreviations: DEGs: differentially expressed genes
Immune cell infiltration profilingTo assess the immune microenvironment changes in Lyme disease, we performed immune cell deconvolution analysis using CIBERSORT on the GSE145974 dataset. The relative proportions of 22 immune cell types were quantified across all samples. A correlation heatmap revealed distinct relationships among immune cell populations in LD (Fig. 6a).
Fig. 6
Immune infiltration analysis. a Correlation heatmap of the immune microenvironment in patients with LD, b bar plot comparing the expression of immune cells between patients with LD and controls, c box plot comparing the expression of immune cells between patients with LD and controls, d correlation between infiltrating immune cells and FCGR1B/MPP1 expression. Significance levels are indicated as follows: *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001
Monocytes and M0 macrophages were significantly elevated in LD samples compared to controls, while resting CD4 memory T cells were markedly decreased (Fig. 6b–c). These changes suggest an enhanced innate immune state and a possible suppression or redistribution of adaptive T-cell populations. Such an immune profile is consistent with early host responses to bacterial infection.
Moreover, correlation analyses demonstrated that expression levels of FCGR1B and MPP1 were positively correlated with the abundance of monocytes (Fig. 6d). This further supports the functional role of these genes in shaping the innate immune response to Borrelia infection. Collectively, these findings suggest that Lyme disease is characterized by a restructured immune landscape, featuring monocyte/macrophage activation and adaptive immune modulation.
Comments (0)