
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Remember me
The Gaussian network model (GNM) has been successful in explaining protein dynamics by modeling proteins as elastic networks of alpha carbons connected by harmonic springs. However, its uniform interaction assumption and neglect of higher-order correlations limit its accuracy in predicting experimental B-factors and residue cross-correlations critical for understanding allostery and information transfer. This study introduces an information-theoretic enhancement to the GNM, incorporating mutual information-based corrections to the Kirchhoff matrix to account for multi-body interactions and contextual residue dynamics. By iteratively optimizing B-factor predictions and applying a Monte Carlo-driven maximum entropy approach to refine covariances, our method achieves significant improvements, reducing RMSDs between predicted and experimental B-factors by 26%–46% across nine representative proteins. The model contextualizes residue assignments based on local density, solvent exposure, and allosteric roles, showing complex dynamic patterns beyond simple neighbor counts. Enhanced predictions of mutual information and entropy perturbations in proteins like KRAS improve the identification of spanning trees containing key residues, which may correspond to allosteric communication pathways. This evolvable framework, capable of incorporating additional effects and utilizing contextual residue assignments, enables precise studies of mutation effects on protein dynamics, with improved cross-correlation predictions potentially increasing accuracy in drug design and function prediction.
Export citation and abstractBibTeXRIS
The Gaussian network model (GNM) [1] emerged from the theoretical framework of polymer physics, specifically drawing upon the constrained junction theory of rubber elasticity pioneered by Flory [2] and later refined in the Flory-Erman constrained junction model (CJM) [3]. This intellectual lineage, transferring concepts from synthetic polymer networks to biological macromolecules, provided crucial insights into the relationship between molecular topology and conformational dynamics. By conceptualizing folded proteins as elastic networks with alpha carbons serving as junction points connected by fictitious springs, the GNM successfully captured the essence of protein fluctuations governed by local crowding effects.
While the original formulation of the GNM has proven effective in predicting experimental B-factors using just a single adjustable parameter, it showed the possibility of rapidly predicting cross-correlations of fluctuations through the inversion of the Kirchoff matrix, a critical advancement for understanding allostery, information transfer pathways, and protein function more generally. This predictive capability has been particularly valuable, as direct experimental observation of such cross-correlations is only now emerging, with available data still relatively scarce [4, 5].
Systematic deviations in loop regions and densely packed structural elements have been observed when comparing GNM predictions with experimental B-factors, pointing to limitations in the current formulation of the Kirchhoff matrix. Addressing these discrepancies is critical for obtaining more accurate B-factor predictions, which would consequently lead to more reliable cross-correlation estimations that better capture true protein dynamics. While all-atom molecular dynamics (MDs) simulations can provide such detailed correlations, the coarse-grained GNM offers a significantly faster, albeit approximate, computational approach to obtaining this essential functional information.
This paper presents an information-theoretic approach to enhancing the GNM by systematically correcting the Kirchhoff matrix elements to minimize discrepancies between experimental and predicted B-factor profiles. The method uses experimental B-factors as reference data and iteratively modifies each element of the Kirchhoff matrix based on conditional mutual information that quantifies the effect of specific residue interactions on B-factor prediction accuracy. The approach involves partitioning residue sets that contribute to different orders of conditional mutual information terms, with each residue’s inclusion in a particular set determined by whether or not it reduces the RMSD between experimental and GNM-predicted B-factor profiles. Through this greedy optimization procedure using iterative acceptance/rejection, the method first reconstructs a more accurate Kirchhoff matrix that optimally fits the experimental B-factor data. Once this optimization is complete, the approach then maximizes entropy to ensure the least biased solution while keeping the experimentally determined B-factors fixed. Rather than predicting B-factors de novo, this approach utilizes experimental data to derive a corrected covariance structure that shows detailed allosteric pathways, functional dynamics, and long-range correlations that are otherwise inaccessible from experimental B-factor measurements alone.
1.1. Theoretical frameworkAn elastic polymeric network consists of chains linked to each other by covalent junctions. Alternatively, such a network may be conceptualized as a single long primary chain folded in three-dimensional space and covalently connected to itself at distinct points forming junctions. Each junction fluctuates within a coordination sphere defined by a specific cutoff radius, where approximately 50–100 other junctions may be present, many originating from topologically distant segments. Consequently, junction fluctuations are governed by both network connectivity and the degree of interpenetration within this coordination sphere [3, 6]. The mean-square fluctuation of each junction exhibits an inverse relationship with the total number of neighboring junctions, both covalently bonded and non-bonded, that interpenetrate the coordination sphere. The statistical mechanical framework of the CJM has demonstrated remarkable success in explaining virtually all observed behaviors of polymeric elastomers [7–12].
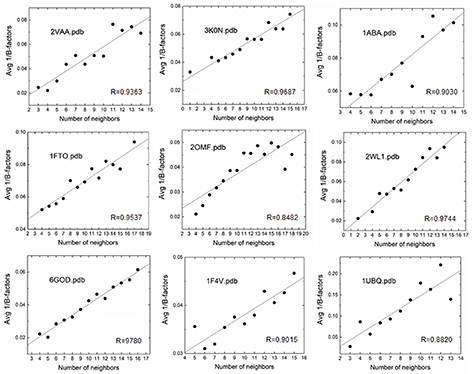
The GNM extends this paradigm to proteins, with alpha carbons assuming the role of junctions [1]. Each alpha carbon interacts with two types of neighbors: those covalently connected along the primary chain and those originating from topologically distant segments. Every alpha carbon occupies a well-defined equilibrium position around which it undergoes thermal fluctuations. Thus, analogous to elastic networks, proteins function as deformable elastic bodies whose elasticity is predominantly determined by these fluctuations. Following the CJM approach, the GNM mathematically implements the physical principle that the mean squared fluctuation of an alpha carbon varies inversely with the total number of neighboring alpha carbons, both covalent and non-covalent. The validity of this physical principle is shown in figure 1 for several examples.
Figure 1. Inverse relationship between B-factors and neighbor counts. This scatter plot displays the relationship between neighbor counts (number of alpha carbons within a 7.8 Å radius) and inverse B-factors (1/B-factor, Å−2). Each data point represents the mean B-factor for residues with a specific neighbor count. A least-squares regression line is fitted to the data, illustrating a linear trend where residues with fewer neighbors (sparse) exhibit higher B-factors, and those with more neighbors (crowded) show lower B-factors. Pearson coefficients, R, for the linear fit are shown in each figure. The data points are obtained by extracting B-factors for each alpha carbon atom and determining the number of neighboring residues within a 7.8 Å radius from the protein data bank structure. For each neighborhood size, the mean B-factor is calculated and plotted. Complete datasets showing individual B-factor and neighbor count values are provided in supplementary figure S-0, with corresponding correlation coefficients (R) and determination coefficients (R2) displayed in each panel.
Download figure:
Standard image High-resolution imageFigure 1 supports Flory’s hypothesis that residues in crowded regions fluctuate less, extending beyond elastic polymeric networks to describe coarse-grained protein behavior. The observed scatter in the figure reflects that while neighbor count provides a simple metric of local packing density, B-factors incorporate numerous additional contributions including, but not limited to, secondary structure elements, solvent accessibility, allosteric and nonlinear dynamic effects, complex factors that cannot be captured by neighbor counts alone.
This completes the analogy wherein a protein is represented as a coarse-grained polymer chain folded onto itself, with alpha carbons fluctuating about their respective equilibrium positions. Both CJM and GNM assume multivariate Gaussian distributions for these fluctuations, resulting in linear spring-like forces between pairs. MDs simulations largely validate this assumption, demonstrating that protein fluctuations indeed approximate Gaussian behavior, consistent with expectations from the central limit theorem [13].
Paul Flory’s groundbreaking insight explained the inverse relationship between junction fluctuations and the degree of interpenetration from neighboring junctions in elastic networks. This fundamental observation, underappreciated at its inception [14], exemplifies Flory’s extraordinary scientific foresight. The principle has transcended its original context to become foundational in our understanding of protein dynamics through the GNM, establishing a crucial bridge between polymer physics and structural biology.
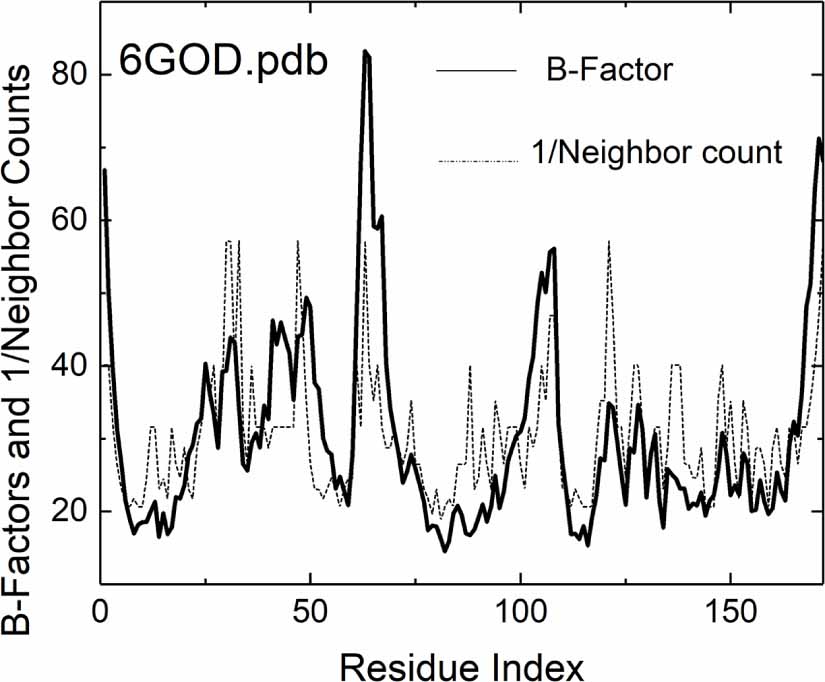
Figure 2 further illustrates the effect of crowding on fluctuations in the widely studied protein KRAS (PDB ID: 6GOD) by comparing experimentally observed B-factor profiles (solid line) with the inverse of alpha carbon neighbor counts for each residue (dotted line), though this simplified metric excludes the influence of network connectivity present in the full GNM. The dotted line, derived simply by calculating the reciprocal of the number of neighboringalpha carbons (degree of interpenetration) and least-squares fitted to experimental B-factor profile, demonstrates the significant contribution of local crowding to residue fluctuations. Notably, the dashed curve reflects only local neighbor effects; contributions from connectivity, represented by the inverse Kirchhoff matrix, are not incorporated. The GNM extends this local crowding effect by integrating the protein’s complete interaction network (to be compared below in figure 5), generalizing the Kirchhoff matrix approach from phantom network theory [15, 16] to encompass both covalent and non-covalent interactions.
Figure 2. Comparison of experimental B-factors (solid lines) with the crowding effect (dotted lines). The crowding effect is calculated from the pdb structure 6GOD.pdb by counting the alpha carbon neighbors within a 7.8 Ångstrom radius for each alpha carbon, taking the reciprocal of this count, and then fitting it to the experimental B-factor profile using least squares. Network connectivity effects as obtained from the inversion of the Kirchoff matrix are not included in this comparison. RMSD of the fit is 10.29. Inclusion of network connectivity effects using the Kirchhoff matrix of the original GNM decreases this RMSD to 8.33. The RMSD using the extended GNM further decreases the value to 5.09. (See figure 5 below). (See table 1 for the RMSD values of the other proteins).
Download figure:
Standard image High-resolution imageComparative analysis between GNM predictions and experimental B-factor profiles shows that despite its general efficacy, GNM systematically underpredicts loop or turn fluctuations while overpredicting fluctuations in densely packed regions. The underestimation of loop mobility can be attributed to several factors. First, the harmonic interaction assumption imposes a quadratic energy landscape, whereas loops often exhibit large-amplitude, anharmonic motions, particularly in flexible or disordered regions. Second, GNM’s fixed cutoff distance (typically 7–10 Å) for residue connectivity may inadequately capture the sparse contact patterns characteristic of extended loops. Third, long-range interactions become proportionally more significant in these sparsely connected regions, as they face less competition from the numerous short-range contacts that dominate in densely packed structural elements. Fourth, solvent exposure increases the effective flexibility of loop residues by reducing the constraining effects of the protein matrix, while GNM treats all residues within the cutoff distance equivalently regardless of their burial state. Most significantly, loop dynamics are substantially influenced by entropic effects as these regions sample multiple conformational states, an aspect not explicitly addressed in the harmonic GNM framework. Conversely, the over prediction of fluctuations in crowded regions suggests insufficient representation of multibody effects within GNM’s mathematical structure. A residue in the crowded environment of its neighbors may experience three-body effects where a third residue may interfere with the interaction of i and j. Four-body effects should also be considered where two residues k and l modify the neighborhood of i and j. These effects will be deeply affected by the size and type of side-chains and will modify the backbone coupling, electronic interactions and long-range correlations that emerge from the entire protein structure. The GNM has no context beyond counting number of neighbors and incorporating effects of chain connectivity on B-factors, a limitation originating from its theoretical roots in CJM of rubber elasticity, where interpenetration of junctions provides sufficient description for homogeneous polymeric systems, but becomes inadequate for proteins where residue diversity and specific interaction patterns demand contextual treatment. However, as noted above, B-factors reflect a composite of factors beyond neighbor and connectivity effects: secondary structure, solvent exposure, and sequence context, dynamic heterogeneity in the protein and multiple conformations and disorder. B-factors are the experimental outcome of these effects. A realistic model needs to be contextualized by systematically addressing these additional factors but it should contain the essential details avoiding unnecessary complexity yet be evolvable. An information-theoretic approach offers a promising avenue, employing concepts such as mutual information conditioned on the presence of several alpha carbons within the coordination shell of a given alpha carbon. This conceptual framework enables us to incorporate conditional dependencies and higher-order correlations such as three-body and higher correlations that may be crucial for accurately modeling the complex dynamics of protein structures.
The simplest refinement of the GNM is to keep the structure of the connectivity matrix (the Kirchhoff matrix) unchanged while modifying the nature of the interactions and including those effects beyond network connectivity and interpenetration. This will modify the interpretation of the fictitious spring concept, which is rooted in the statistical mechanics of a Gaussian polymer chain, which fluctuates under forces applied to its ends [16]. The modification of the Kirchoff matrix should however be made in a self-consistent manner: The zero sum of each row should be retained to ensure equilibrium, the symmetry should be retained to preserve microscopic reversibility or detailed balance, and since every modification of the matrix also modifies its inverse covariance matrix, the iteration of this cycle should converge [17].
While our proposed method offers an approach to predict protein correlations with improved accuracy, the primary aim of this paper is to enhance the GNM by addressing factors not considered in its original formulation. We improve both the accuracy of B-factor predictions and the evaluation of covariances, the off-diagonal terms of the Kirchhoff inverse, in a self-consistent manner following the principles of information theory. Although accurate B-factor assessment is essential for understanding protein dynamics, proper evaluation of covariances is even more critical, as key functional mechanisms, such as allosteric signaling and energy transfer, propagate through residue correlations. Covariances are presently widely studied through time-intensive MDs simulations [18], with direct experimental determination only recently emerging [4, 5], making our computationally efficient approach particularly timely.
The primary contributions of this work are threefold. First, we demonstrate significantly improved agreement between predicted and experimental B-factors compared to the standard GNM. Second, we introduce a framework for contextualizing residue interactions within the GNM, enabling a more accurate representation of the complex correlation patterns that underlie protein function and allostery. Third, we develop a Monte Carlo approach to identify cross-correlations that satisfy the maximum entropy (MaxEnt) principle while maintaining the optimized B-factors as fixed, thereby providing new insights into how information is transferred within protein structures and how distant sites communicate to regulate function.
2.1. The GNM and the formulation of the extended modelThe ij’th element of the Kirchoff matrix of the original GNM is defined as
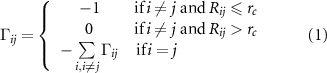
where 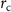 is the cutoff separation defining the range of interaction of α carbons, and
is the cutoff separation defining the range of interaction of α carbons, and 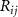 is the distance between the ith and jth
is the distance between the ith and jth 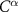 atoms. The presence of −1 at position (i, j) signifies the influence of residue j on the fluctuations of i. Since the interaction is inherently symmetric, the influence of i on j is identical, ensuring that the Kirchhoff matrix remains symmetric.
atoms. The presence of −1 at position (i, j) signifies the influence of residue j on the fluctuations of i. Since the interaction is inherently symmetric, the influence of i on j is identical, ensuring that the Kirchhoff matrix remains symmetric.
In the GNM, interactions lack context, with all contacts assumed to be identical and modeled solely as uniform pairwise interactions. However, pairwise interactions in proteins depend on multiple factors, including steric interactions, hydrogen bonds, and electrostatic forces, which introduce variability not captured by this simplification. Furthermore, the GNM overlooks higher-order interactions, such as triplet contributions, where the combined effect of neighbors j and k influences the fluctuations of residue i, or quadruplet interactions involving neighbors j, k, and l, which may also be modulated by these factors. Given that the interaction domain of a residue i may include up to 15 neighbors, these triplet and higher-order interactions could be significant. For instance, the combined influence of residues k and l can alter the information transfer between residues i and j, effectively changing how strongly these residues are coupled through the network. Such effects are not accounted for in the GNM, which thus serves as a zeroth-order approximation to the more refined model presented below.
The extended model retains the framework of the GNM while accounting for modifications to the i,j-potential based on the effects outlined in the preceding paragraph and explained in more detail in the following paragraphs. Specifically, the interaction between residues i and j, beyond the uniform GNM potential, is adjusted to reflect the influence of other residues within the interaction domain of i and j. These modifications are incorporated into the i,j-th element of the Kirchhoff matrix, 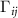 , capturing the contextual and multiplet contributions to the pairwise dynamics as follows:
, capturing the contextual and multiplet contributions to the pairwise dynamics as follows:
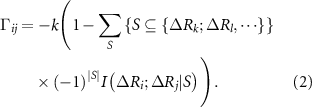
Here, k is the spring constant of the original GNM theory, the 1 in the parenthesis represents the spring force in the original GNM, and the sum represents the conditional mutual information between the fluctuations of residue i and j, systematically accounting for the effects of conditioning on the variables k, l, … The summation runs over all subsets S of the set 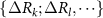 , meaning it considers all possible ways to condition on none, one, both, etc., of these variables.
, meaning it considers all possible ways to condition on none, one, both, etc., of these variables. 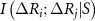 denotes the mutual information between the fluctuations of residues i and j while conditioning on the subset S. The factor
denotes the mutual information between the fluctuations of residues i and j while conditioning on the subset S. The factor 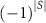 alternates the sign depending on the number of elements in S. This ensures proper cancellation of redundant contributions, following the principle of inclusion–exclusion [19]. This correction represents a mean-field type adjustment, averaged over the fluctuations of all remaining residues k,l,… and will be explained in more detail below.
alternates the sign depending on the number of elements in S. This ensures proper cancellation of redundant contributions, following the principle of inclusion–exclusion [19]. This correction represents a mean-field type adjustment, averaged over the fluctuations of all remaining residues k,l,… and will be explained in more detail below.
The summation term in equation (2) represents the contributions of non-GNM effects to  . Inversion of this modified
. Inversion of this modified  leads to the covariance of fluctuations, which is then used to obtain the new Kirchhoff matrix
leads to the covariance of fluctuations, which is then used to obtain the new Kirchhoff matrix  . We demonstrate that iterative application of this equation converges rapidly. At each step of the iteration, the
. We demonstrate that iterative application of this equation converges rapidly. At each step of the iteration, the  matrix evolves from the unweighted GNM representation to a dynamically reweighted interaction matrix that integrates information-theoretic corrections based on mutual information.
matrix evolves from the unweighted GNM representation to a dynamically reweighted interaction matrix that integrates information-theoretic corrections based on mutual information.
The diagonal elements of  yield variances proportional to experimentally determined B-factors, providing a basis for validating the model’s accuracy. More importantly, the off-diagonal elements of
yield variances proportional to experimentally determined B-factors, providing a basis for validating the model’s accuracy. More importantly, the off-diagonal elements of 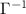 show correlations between residue fluctuations. Since
show correlations between residue fluctuations. Since 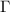 is singular, its inverse is obtained as the Moore–Penrose pseudoinverse, shown as
is singular, its inverse is obtained as the Moore–Penrose pseudoinverse, shown as 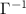 or equivalently as
or equivalently as 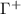 throughout the paper. Since the inverse operator generates correlations between spatially distant residues, an accurately constructed
throughout the paper. Since the inverse operator generates correlations between spatially distant residues, an accurately constructed 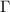 is crucial for understanding allostery and information/entropy transfer in proteins.
is crucial for understanding allostery and information/entropy transfer in proteins.
 matrix
matrixThe summand in equation (2) expands to:
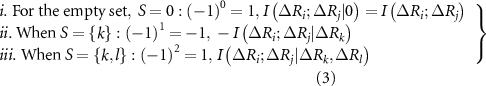
where, 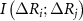 ,
, 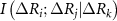 , and
, and 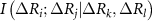 are the joint mutual information between residues i and j, the mutual information between i and j after accounting for the influence of residue k, and the mutual information after accounting for the joint effect of residues k and l, respectively. We consider only up to four body interactions. Using equation (3) in equation (2) and properly scaling each term we obtain
are the joint mutual information between residues i and j, the mutual information between i and j after accounting for the influence of residue k, and the mutual information after accounting for the joint effect of residues k and l, respectively. We consider only up to four body interactions. Using equation (3) in equation (2) and properly scaling each term we obtain
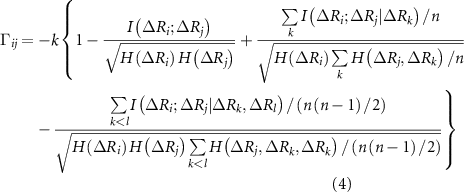
where, 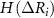 ,
, 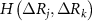 and
and 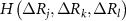 are the singlet, doublet an triplet entropies and n is the number of neighbors of i. This systematically accounts for the information between residues i and j while properly including and excluding the effects of residues k and l. The alternating signs ensure that there is no double-counting information and are correctly accounting for how k and l individually and jointly affect the i–j interaction. In equation (4), in order to align with GNM’s unit contact strength −1,
are the singlet, doublet an triplet entropies and n is the number of neighbors of i. This systematically accounts for the information between residues i and j while properly including and excluding the effects of residues k and l. The alternating signs ensure that there is no double-counting information and are correctly accounting for how k and l individually and jointly affect the i–j interaction. In equation (4), in order to align with GNM’s unit contact strength −1, 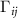 is refined using averaged entropy terms,
is refined using averaged entropy terms, 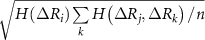 and
and 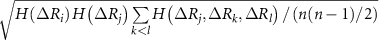 . These factors ensure adjustments that are commensurate with the GNM’s unit contact strength (−1), typically yielding values below 1, which preserves matrix stability and avoids over-amplification. An alternative normalization to [0, 1] using
. These factors ensure adjustments that are commensurate with the GNM’s unit contact strength (−1), typically yielding values below 1, which preserves matrix stability and avoids over-amplification. An alternative normalization to [0, 1] using 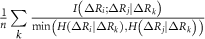 and
and 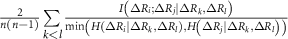 was tested but introduced negative eigenvalues, confirming the original scaling’s suitability.
was tested but introduced negative eigenvalues, confirming the original scaling’s suitability.
Equation (4) will be evaluated iteratively where the zeroth iteration will be the original GNM. The mutual information terms are functions of the covariance of the fluctuations. The covariance obtained by inverting  will be used to obtain the new mutual information values, which will be used on the right hand side in the next iteration. Iterations of equation (4) give the modified Kirchhoff matrix.
will be used to obtain the new mutual information values, which will be used on the right hand side in the next iteration. Iterations of equation (4) give the modified Kirchhoff matrix.
To choose Kirchhoff matrix elements in the extended GNM, we construct three complementary sets:
Set 0 (enhanced two-body interactions): This set, represented by interactions of the form 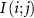 , encompasses residues where residue i is directly affected by residue j, similar to the original GNM with Kirchhoff matrix coefficients of −1. The extended GNM uses the same distance cutoff as the standard GNM for these interactions.
, encompasses residues where residue i is directly affected by residue j, similar to the original GNM with Kirchhoff matrix coefficients of −1. The extended GNM uses the same distance cutoff as the standard GNM for these interactions.
Set 1 (three-body crowding interactions): This set captures interactions of the form 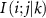 , representing crowded environment effects where residue k in the neighborhood constrains the mobility of residue i. These terms carry opposite signs to set zero interactions, reflecting that the presence of residue k decreases the fluctuations of residue i by creating a more restrictive local environment. This represents a three-body interaction that accounts for crowding effects on residue dynamics.
, representing crowded environment effects where residue k in the neighborhood constrains the mobility of residue i. These terms carry opposite signs to set zero interactions, reflecting that the presence of residue k decreases the fluctuations of residue i by creating a more restrictive local environment. This represents a three-body interaction that accounts for crowding effects on residue dynamics.
Set 2 (four-body interactions): This set includes interactions 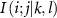 where two neighboring residues k and l collectively influence the i–j interaction. We focus specifically on residues that exhibit increased B-factors, as these represent sites where the standard GNM underpredicts flexibility. The four-body interaction term captures complex cooperative effects where neighboring flexible residues collectively enhance the mobility of the i–j pair. This approach is justified because: (1) the primary goal is to correct systematic underpredictions in flexible regions, and (2) these cooperative mobility enhancements are most pronounced in sparse environments where residues have greater conformational freedom, as opposed to densely packed regions where motion is already constrained and additional coupling changes have minimal impact.
where two neighboring residues k and l collectively influence the i–j interaction. We focus specifically on residues that exhibit increased B-factors, as these represent sites where the standard GNM underpredicts flexibility. The four-body interaction term captures complex cooperative effects where neighboring flexible residues collectively enhance the mobility of the i–j pair. This approach is justified because: (1) the primary goal is to correct systematic underpredictions in flexible regions, and (2) these cooperative mobility enhancements are most pronounced in sparse environments where residues have greater conformational freedom, as opposed to densely packed regions where motion is already constrained and additional coupling changes have minimal impact.
The division into three sets is based on the physical insight that different local environments require different interaction models. Set 0 captures standard pairwise interactions similar to the original GNM. Set 1 addresses crowding effects where additional neighbors constrain mobility through three-body interactions. Set 2 captures cooperative flexibility enhancement in sparse environments where neighboring flexible residues collectively increase mobility through four-body interactions. This classification reflects the fundamental principle that residue dynamics depend not only on direct contacts but also on the local structural context.
Using several proteins, we found that partitioning of residues into these sets is governed by three key factors: (1) neighborhood count, (2) solvent accessible surface area (SASA), and (3) dynamic coupling. The dynamic coupling factor accounts for long-range communication, allosteric effects, and entropy perturbability. We describe the evaluation of these factors in detail in section 2.3.1 below.
Residue contextualization was performed using a score defined as:

with equal weights w1 = w2 = w3 = w4 = 1 and 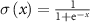 is the sigmoid function. The indices min and range denote the minimum value of the given factor and the difference between maximum and minimum, respectively.
is the sigmoid function. The indices min and range denote the minimum value of the given factor and the difference between maximum and minimum, respectively.
Residues with scores between 0.5 and 1 are assigned to either Set 0 or Set 2 (increased flexibility, higher B-factors) during the calculations, while residues with scores between 0–0.5 are assigned to Set 1 (decreased flexibility, lower B-factors). The assignment procedure operates as follows: residues are first classified as either List 1 (B-factor increasing) or List 2 (B-factor decreasing) according to their scores. Each residue is then iteratively assigned to its designated set, with the RMSD between predicted and experimental B-factors calculated after each assignment. If the RMSD decreases with the assignment, that residue is permanently assigned to its designated set. This process continues until all residues from both lists have been assigned to their respective sets.
One potential limitation of this approach is that residues initially misclassified into the wrong list cannot subsequently find their correct assignment. However, such misclassifications are most likely to occur at the boundary between List 1 and List 2 residues, where the sigmoidal classification function transitions (at the 50th percentile threshold). To assess this potential source of error, random checks were performed on borderline residues, showing classification errors in less than 1% of cases. This error rate produces negligible differences in the final predicted B-factors.
We tested assigning all residues to Set 2 (four-body interactions) for 6GOD. This resulted in computational instability and RMSD degradation to 12.4 Ångström2 (vs 5.09 for optimized assignment). The failure occurs because four-body terms are physically meaningful only in sparse, flexible environments. Applying them universally introduces artificial correlations in constrained regions where such interactions are physically unrealistic and mathematically destabilizing.
2.3.1. Evaluation of number of neighbors, SASA, and dynamic coupling for individual residues2.3.1.1. Neighborhood countQuantifies the local density of contacts around each residue, reflecting constraints on mobility. Based on the Flory hypothesis, this factor correlates with experimental B-factors, with protein 6GOD.pdb serving as a representative example in figure 2. Agreement parameters for all proteins studied are reported in the second column of table 1. Residues with higher contact counts typically exhibit lower B-factors due to increased structural constraints.
Table 1. Deviation (RMSD) of the original and the extended GNM from experimental B-factors.
ProteinRMSD 1/B-NEIGHBORRMSD GNMRMSD extended GNMPercent improvement over GNM6GOD10.298.335.0938.91F4V5.905.843.6138.21ABA3.243.171.8043.21FTO4.934.543.0632.62OMF13.0012.657.2442.82VAA10.248.824.7446.32WL114.3814.0510.4625.63K0N4.394.273.1226.92NMO2.502.291.6926.22.3.1.2. SASACalculated using the Shrake-Rupley algorithm [20]. The method approximates a water molecule (probe radius 1.4 Å) rolling over the protein surface to determine accessible surface points, summing atomic contributions to yield per-residue SASA. Expressed as percent solvent accessibility (%SAS), higher SASA indicates surface-exposed residues with fewer contacts, leading to greater mobility and higher B-factors. Buried residues (low SASA) are more constrained, resulting in lower B-factors.
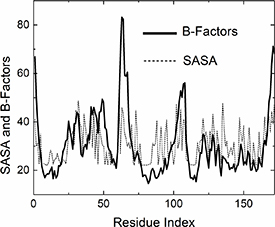
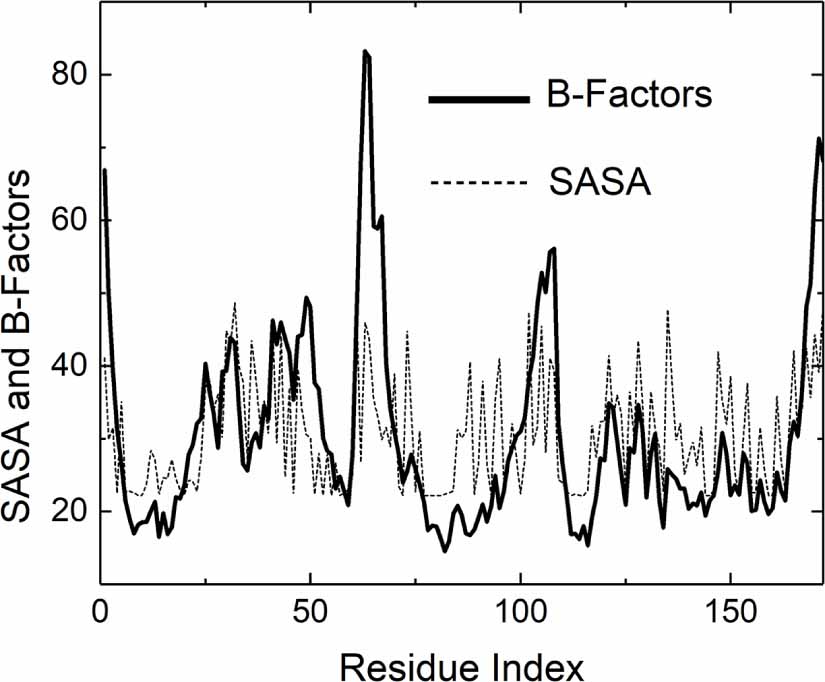
The reasonable agreement between B-factor and SASA profiles in figure 3 occurs because both reflect similar underlying structural principles: (i) surface exposure and mobility. Residues with high SASA are typically on the protein surface and have fewer constraining contacts with neighboring residues. This reduced constraint allows for greater thermal fluctuations, leading to higher B-factors. Conversely, buried residues (low SASA) are more constrained by their local environment, resulting in lower B-factors. Both metrics capture the degree of structural constraint experienced by each residue. SASA directly measures solvent accessibility (indicating surface vs buried locations). B-factors reflect the dynamic consequence of these constraints (more constrained = less mobile).
Figure 3. Comparison of B-factor and SASA profiles for 6GOD.pdb. The solid curve represents experimental B-factors, while the dashed curve shows SASA (calculated by the Shrake-Rupley algorithm [20] and scaled for least squares fit to B-factor profile). Peaks and minima align, reflecting shared structural principles: surface exposure correlates with higher mobility and B-factors, while buried residues show reduced dynamics.
Download figure:
Standard image High-resolution imageThis correlation suggests that solvent accessibility is a key determinant of residue mobility, making SASA a useful predictor for understanding B-factor patterns in protein structures.
2.3.1.3. Dynamic coupling: the third factor quantifies the dynamic coupling between residues using the metric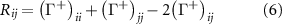
where 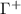 is the Moore–Penrose pseudoinverse of the Kirchhoff matrix. This quantity represents the mean-square fluctuation correlation
is the Moore–Penrose pseudoinverse of the Kirchhoff matrix. This quantity represents the mean-square fluctuation correlation 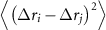 between residues i and j, measuring their dynamic coupling. For comparison with experimental B-factors
between residues i and j, measuring their dynamic coupling. For comparison with experimental B-factors 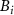 , we define the cumulative dynamic coupling of residue i as
, we define the cumulative dynamic coupling of residue i as
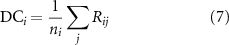
where 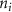 is the number of contacts of residue i. In supplementary material sections S-4 and S-5 we demonstrated that
is the number of contacts of residue i. In supplementary material sections S-4 and S-5 we demonstrated that 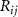 is fundamentally linked to entropy perturbability through the relation
is fundamentally linked to entropy perturbability through the relation
Comments (0)