
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Remember me
Phenotypic plasticity, the ability of cells to change their phenotypes reversibly in response to changing environments, is a fundamental phenomenon (Mills et al 2019) across different contexts, including embryonic development and disease (Jopling et al 2011, Gupta et al 2019, Kalra et al 2021, Pérez-González et al 2023). Cell-state transitions often require a large-scale change in the transcriptomic profile, which are governed by the underlying gene regulatory networks (GRNs). Some topological features of GRNs, such as feedback and feedforward loops, have a significant impact on the dynamics of phenotypic plasticity (Murphy et al 2002, Zhang et al 2016, Hebbar et al 2022). Many embryonic cell-fate transitions are governed by relative expression levels of two mutually repressing genes (Zhou and Huang 2011). This regulatory motif of two genes (A and B) mutually inhibiting each other is called a toggle switch (TS) and can give rise to two co-existing stable steady states that can switch back and forth: (high A, low B) and (low A, high B) (Gardner et al 2000). For example, the master regulator genes PU.1 and GATA1 form a TS that influences the differentiation of common myeloid progenitor cells into myeloid and erythroid progenitors based on the relative expression of these two genes. (High PU.1, low GATA1) facilitates the differentiation of the common myeloid progenitor cell into myeloid cells, while (low PU.1, high GATA1) leads to differentiation into erythroid cells. A third state, enabled by self-activation on the two transcription factors (TFs), has a balanced expression level of both PU.1 and GATA1 molecules and corresponds to the progenitor state (Roeder and Glauche 2006). Similar binary cell-fate decision systems are also important in cancer (Sahoo et al 2021, Pérez-González et al 2023). A prime example is epithelial-to-mesenchymal transition (EMT), where a TS between the family of TFs ZEB1/2 and the family of miR-200 microRNAs has been identified to be a core motif in the underlying GRNs (Brabletz and Brabletz 2010, Celià-Terrassa et al 2018). The self-activation of ZEB1 supports the emergence of a hybrid E/M state, composed of expression of both ZEB1/2 and miR200 molecules, similar to the progenitor state in the (PU.1, GATA1) example (Lu et al 2013, Jolly et al 2018). These hybrid E/M states are more stem-like, drug-resistant, and metastatically fit (Pastushenko and Blanpain 2019).
The dynamics of a TS, including TFs and/or microRNAs, have been well-studied using deterministic and stochastic models to capture qualitative features of binary cell-fate decisions (Roeder and Glauche 2006, Tian and Burrage 2006, Shiraishi et al 2010, Jaruszewicz and Lipniacki 2013, Okawa et al 2016). Furthermore, the emergent properties of the TS are resilient to external perturbations, making the TS a robust cell-fate decision module (Harlapur et al 2022). However, across different contexts, the underlying GRNs of cell-fate decisions have a larger number of nodes, encompassing multiple TSs and feedback loops (Ríos et al 2015, Udyavar et al 2017, Hari et al 2020, Arthur et al 2024). Recently, we analyzed the networks governing EMT and found the presence of two mutually inhibiting teams of nodes (Hari et al 2022b). A team is a group of genes that activate member genes belonging to the same team while inhibiting genes of other teams directly and/or indirectly. For instance, in the EMT network, most members of the epithelial team were EMT-inhibiting microRNAs such as miR-200 family, while most members of the mesenchymal team were EMT-inducing TFs (EMT-TFs) such as ZEB1 and SNAI1. These EMT-TFs activated each other and also repressed microRNAs that targeted them or other EMT-TFs, while many microRNAs inhibited EMT-TFs that inhibited them or other microRNAs in the same team. Such ‘teams’ have also been observed in other biological contexts, including developmental decision-making (Chauhan et al 2021, Pillai and Jolly 2021, Sehgal et al 2024, Hari et al 2025). The intuitive framework of mutually inhibiting teams, their consistent influence on network dynamics, and their presence across diverse biological contexts make them an attractive candidate for being a new topological design principle of GRNs. However, the role of network parameters and external perturbations in shaping the dynamics of team-based GRNs remains poorly understood. In particular, their impact on the phenotypic repertoire of decision-making GRNs has yet to be investigated comprehensively.
Here, we assess the potential of teams of nodes as a broadly applicable design principle by examining their occurrence in biological networks and probing how perturbations affect their emergent dynamics. We first analyzed GRNs from the Cell Collective database, which spans a wide range of biological systems and thus exhibited a wide variation in team strength values. To systematically characterize this variability, we introduced a metric called impurity, defined as the fraction of edges inconsistent with an idealized two-team architecture. Strikingly, impurity correlated with statistical properties of the phenotypic landscapes of Cell Collective GRNs. Building on this insight, we further investigated how impurity, network density, and size shape the dynamics of artificially generated team-based networks. We simulated artificially generated two-teamed networks (TTNs) using two simulation formalisms: primarily a threshold-based Boolean formalism, a discrete state-space approach that considers only the effect of the network topology without any parameter sets (Font-Clos et al 2018), and RACIPE, a continuous state-space, ODE-based simulation formalism that calculates network dynamics over a large parameter space (Huang et al 2018). We quantified two key properties of the phenotypic landscape to evaluate how network parameters shape emergent dynamics: (a) the frequency of pure states (F1)—mutually exclusive states in which one team is ‘on’ and the other ‘off’—and (b) the correlation of expression levels between nodes belonging to opposing teams (CC), using the TS as a reference. By mapping changes in these properties as a function of network parameters, we found consistent trends between artificial networks and those from the Cell Collective database. Our results further demonstrate that TTNs, much like TSs, preserve their dynamical behavior under external perturbations. Using artificial TTNs, we showed that constraining GRNs into a two-team structure enables accurate prediction of experimentally relevant dynamical features, such as node–group correlations and the entropy of steady-state distributions. Together, these findings establish the teams’ paradigm as a powerful framework for linking GRN structure to dynamics. Beyond predictive insights, this paradigm also offers a foundation for developing new methods to infer coarse-grained network properties from high-throughput data, advancing our understanding of emergent cellular behaviors.
2.1. Teams of nodes and binary landscapes in cell-fate decision networksIn this section, we start with a brief introduction to the idea of teams, discussing the network properties, and the properties of the emergent landscapes with examples of cell fate decision networks that we had analyzed previously. Consider the cell-fate decision system—Epithelial–Mesenchymal plasticity (EMP). EMP is a crucial process studied in developmental biology and cancer biology, involving cell sampling phenotypes on a spectrum with epithelial phenotype (E) on one end and mesenchymal phenotype (M) on the other. The phenotypic landscape of EMP, typically representing the abundance of different phenotypes along the EMP spectrum in a population of heterogeneous cells, could be classified as predominantly binary. A salient feature of experimental studies involving EMP is that a heterogeneous cell population primarily would consist of cells with either E or M phenotype, with a small percentage corresponding to hybrid E/M phenotypes. Thus, the phenotypic frequency distribution predominantly looks bimodal, with most of the variance in the state space being explained by a single principal component (Hari et al 2025). This feature of bimodality is observed across a wide variety of cell-fate decision systems, a few examples being NE/non-NE decision system in small cell lung cancer, gonadal cell-fate determination system and a majority of decision systems involved in development. As described in the discussion earlier, such systems could be visualized as being driven by TSs, formed by two master regulators mutually inhibiting each other.
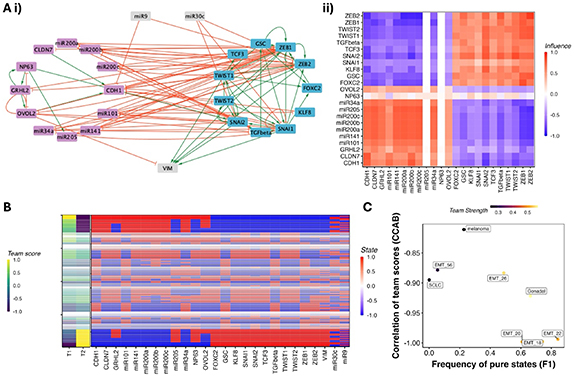
However, we found that the real networks are not as clean-cut. While some networks present high-out degree nodes that could be viewed as the master regulators, other networks do not. In some cases, the markers of a given phenotype do not regulate each other at all, but are co-expressed due to the regulation by the markers of the other phenotype. Despite the individual differences in structure and hierarchy of these networks, we find that they can be united together as networks with teams, by considering not only the direct interactions, but also the indirect interactions. Consider the example in (figure 1(A–i)), an EMP network which has been constructed to explain the phenotypic heterogeneity in a population of cells undergoing EMP. The nodes in the network have been segregated into two groups based on their biological functions: Epithelial and Mesenchymal. Notice how there are no direct interactions between the epithelial nodes (micro-RNAs, labeled as miRs). However, one can realize that the epithelial nodes are inhibiting mesenchymal nodes, and the mesenchymal nodes are, in turn, inhibiting epithelial nodes. Therefore, epithelial nodes are activating themselves indirectly. Inspired by this observation and the fact that the dynamics of a node carry the effect of the regulators of its regulators by proxy, we transform the structure of the network to better represent the dynamics. We integrate the effects of interactions of different path lengths between each pair of nodes, where a path is a series of connected edges between any pair of nodes, and the length of such a path is n−1, where n is the number of nodes in the path. Thus, we can say that while there are no paths of length 1 between Epithelial nodes, there are paths of length 2 mediated by mesenchymal nodes. Following this transformation, we can construct the ‘influence matrix’, which is also a signed, directed graph as the original network, but more connected. This influence matrix can be further clustered to reveal the ‘team’ structure of a network, as shown in figures 1(A–ii) and (S1). The team structure emerges from the network such that the nodes belonging to the same team have a positive influence on each other, while those belonging to the opposite team have a negative influence on each other. It is interesting to note that the influence matrix looks similar to that of a correlation or co-expression matrix, given its connectivity, despite being derived from the underlying regulatory network. Indeed, we were able to show previously that the influence matrix closely resembles the pairwise correlation matrix obtained from simulated steady states of the network (Hari et al 2022b).
Figure 1. Teams and the phenotypic landscape of cell-fate decision networks: (A) Network wiring diagram (i) and the influence matrix (ii) of a 26-node EMT network (Tripathi et al 2020). (B) Concatenated heatmaps depicting the steady state expression levels of the nodes in the EMT network (right) and corresponding team scores of each steady state (left). Each row corresponds to a steady state, and each column corresponds either to a node of the network or a team. The width of each row is proportional to log (frequency). (C) F1 and CCAB of 8 cell fate decision networks. Reproduced from Hari et al (2022b). CC BY 4.0.
Download figure:
Standard image High-resolution imageWe simulated these networks using a threshold-based Boolean formalism (see methods) to obtain an ensemble of steady states that represents the experimental heterogeneous population. The influence matrix thus reveals an underlying toggle-switch with self-activation (TSSA) structure in the network, with each team acting as a single node in TSSA. Importantly, as one would expect from the TSSA, the majority of the state space of the converges to one of the two states where nodes from one team have a high expression and the other team have a low expression (figure 1(B), red-blue matrix—thickness of the row shows the abundance of the steady state)—a binary phenotypic landscape. Using a metric that quantifies the team strength by measuring the strength of influences within and across teams (thereby measuring how well the network allows for a team to act as a single unit, see Methods section 4.2 ), we were able to qualitatively demonstrate that strong teams lead to a strongly bimodal phenotypic landscape. Using the analogy of teams acting as supernodes, we calculated the frequency of pure states (F1, representing the frequency of states with only one team on), which represents the total abundance of the two dominant phenotypes at the ends of the spectrum, and the correlation between the expression levels of the ‘supernode’ (CCAB) calculated for each steady state as the average expression of all nodes in a given team. Networks with higher team strengths showed higher F1 scores and stronger correlations, and vice versa (figure 1(C)). Motivated by these observations, we wanted to ask if we can quantify the effect of network structure on the phenotypic landscape emergent from GRNs.
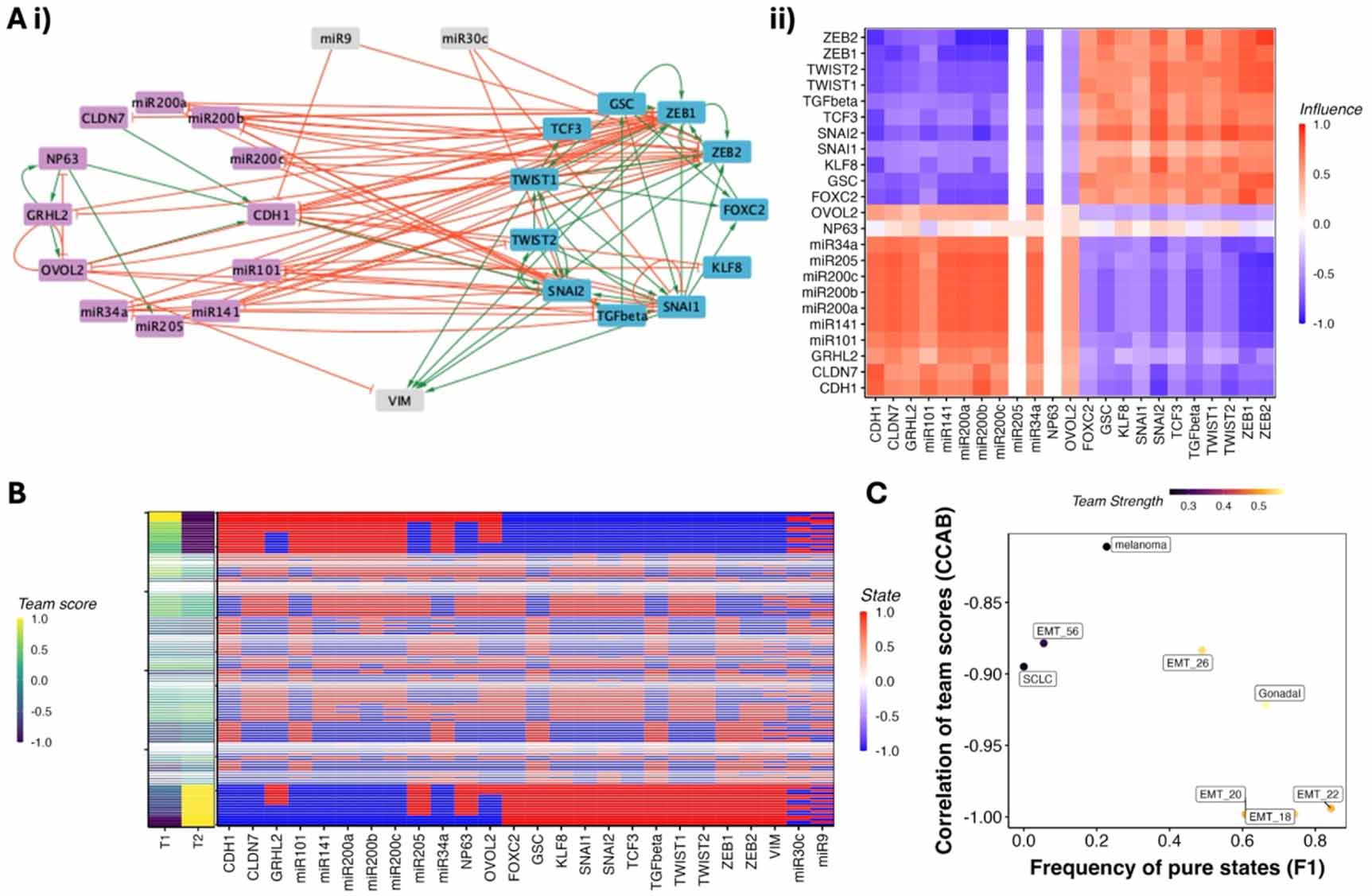
2.2. Network impurity correlates with F1 and CCAB in GRNs from Cell Collective databaseTo test whether the existence of teams is a general feature of GRNs beyond the previously studied cases, we analyzed GRNs from the Cell Collective database (Helikar et al 2012), spanning a range of biological contexts. We eliminated the networks having more than 100 nodes from this analysis for ease of computational tractability, leaving us with 42 networks in total. Of these, approximately 20 networks exhibited mutually inhibiting team structures of varying strengths (see figures 2(A) and S2(A)). In addition, we identified more complex interaction motifs between teams in some networks. For example, some networks exhibited mutually activating teams (e.g. signaling network that include links to propagate and amplify signals through positive feedback loops (der Heyde et al 2014)), while others included negative feedback loops between teams (e.g. cell cycle networks capturing oscillations (Todd et al 2012)). Given the similarity between the influence matrix and the corresponding correlation matrices observed previously, identification of these team-based interaction structures can offer an intuitive lens through which to understand the emergent phenotypic landscape of GRNs. Mutually inhibitory teams tend to result in two distinct, ‘pure’ phenotypic states as shown above: one in which the nodes of one team are predominantly active (expressed), and another in which the opposite is true. On the other hand, mutually activating teams—or, equivalently, a single coherent team—tend to give rise to phenotypes characterized by the co-expression of all team members. The negative feedback was observed in a cell cycle network, where oscillations between cell cycle phases is a desired outcome and can be supported by negative feedback loops (Todd et al 2012). However, we also observed that nearly half of the networks from the Cell Collective database did not exhibit clearly defined teams, and even among those that did, the team structures varied considerably in strength and organization. This observation led us to ask whether it might still be possible to represent these more ambiguous networks using a relaxed concept of ‘impure teams.’
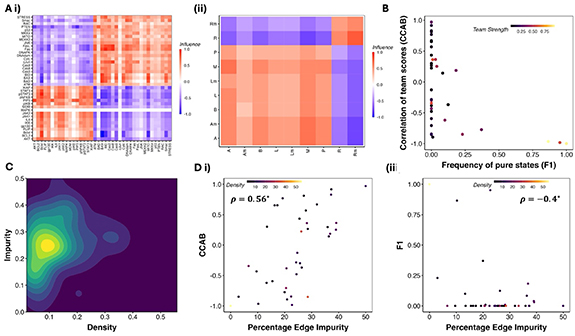
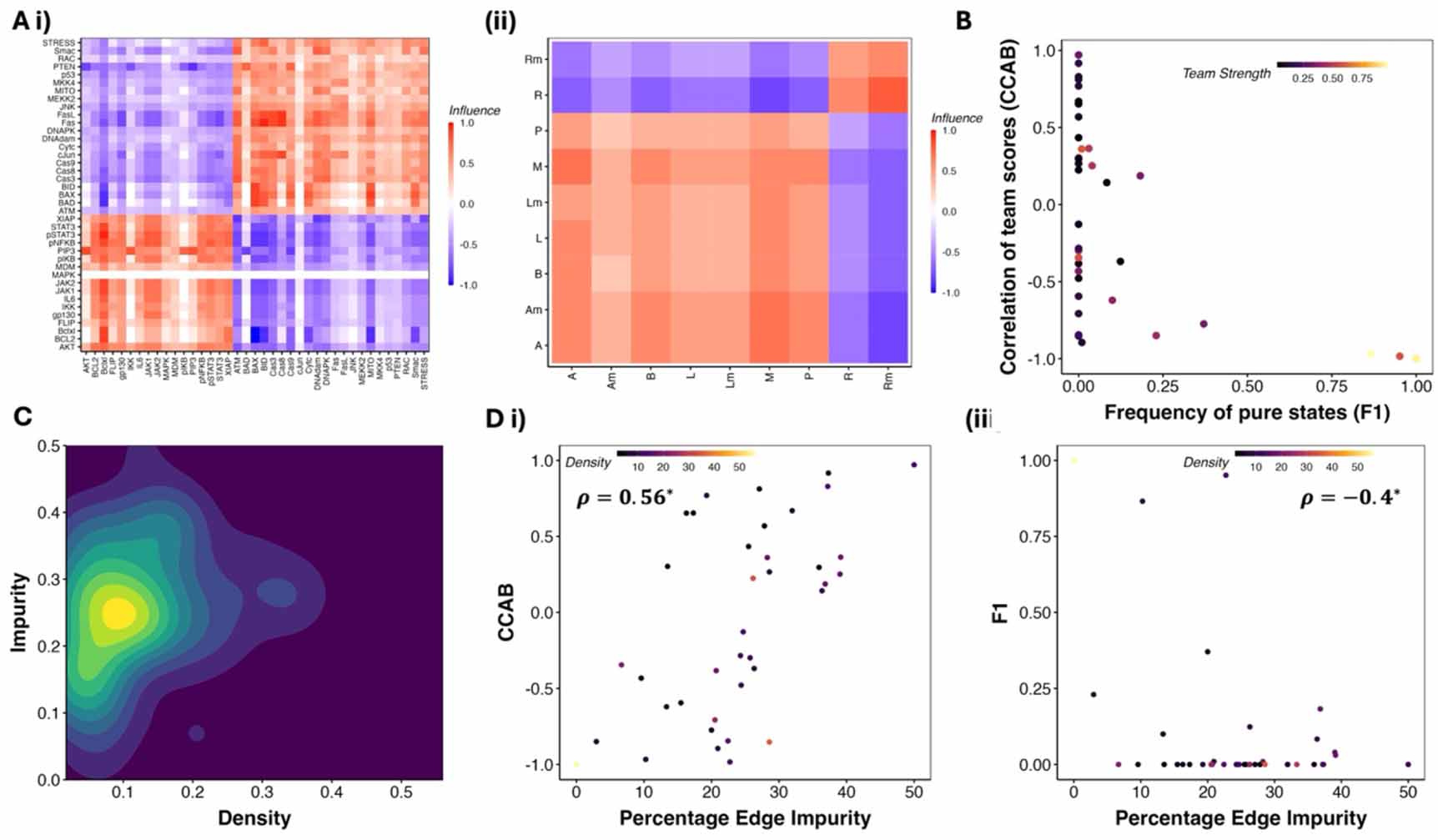
Figure 2. Team-based analysis of GRNS from the Cell Collective database. (A) Examples of Cell Collective GRNS with teams in mammalian (Chudasama et al 2015) and bacterial (Veliz-Cuba and Stigler 2011). (B) F1 and CCAB for Cell Collective networks obtained from simulating the logical rules. Each point corresponds to a network, colored by their team strength. (C) 2D density map of the biological networks depicting the edge density and edge impurity. (D) variations in CCAB and F1 with edge impurity. ρ represents the spearman correlation coefficient, with the * representing statistical significance of the correlation (p-value < 0.05) for a two tailed t test.
Download figure:
Standard image High-resolution imageTo formalize this, we analyzed the influence matrix of each network, defined as the weighted sum of the powers of its adjacency matrix (see Methods 4.1). We then applied clustering algorithms to this matrix to classify the nodes into two provisional teams. Where needed, we refined these classifications manually to best approximate a mutually inhibitory team configuration.
For each network, we quantified two key structural metrics:
1.
Density: defined as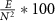 where E is the number of edges and N is the number of nodes in the network.
where E is the number of edges and N is the number of nodes in the network.2.
Impurity: defined as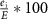 , where
, where 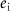 is the number of ‘impure’ edges—those that are inhibitory between nodes within the same team or activatory between nodes in opposing teams. An impurity value of 0% corresponds to a perfect mutually inhibitory team structure, whereas an impurity of 100% indicates a configuration where nodes inhibit their team and activate the opposing team. Biological networks consist of some edges that may appear like impurities in the context of ‘team’ behavior, such as auto-inhibitory feedback loops (for instance, seen in SNAIL1 (Peiró et al 2006)) and incoherent feedforward loops (Lai et al 2016), or mutual inhibition between EMT-TFs such as SNAI1 and SNAI2 (Subbalakshmi et al 2022) or that between ZEB1 and ZEB2 (Vandamme et al 2020). These structures play a crucial role in the contexts of robustness, noise dynamics, and signal transmission (Cinquin and Demongeot 2002, Dublanche et al 2006, Zhang et al 2012). Quantifying the effect of these inconsistent links can be challenging. The ‘teams’ conceptualization thus provides us with a way to quantify the extent of such inconsistencies through the lens of the impurity metric.
is the number of ‘impure’ edges—those that are inhibitory between nodes within the same team or activatory between nodes in opposing teams. An impurity value of 0% corresponds to a perfect mutually inhibitory team structure, whereas an impurity of 100% indicates a configuration where nodes inhibit their team and activate the opposing team. Biological networks consist of some edges that may appear like impurities in the context of ‘team’ behavior, such as auto-inhibitory feedback loops (for instance, seen in SNAIL1 (Peiró et al 2006)) and incoherent feedforward loops (Lai et al 2016), or mutual inhibition between EMT-TFs such as SNAI1 and SNAI2 (Subbalakshmi et al 2022) or that between ZEB1 and ZEB2 (Vandamme et al 2020). These structures play a crucial role in the contexts of robustness, noise dynamics, and signal transmission (Cinquin and Demongeot 2002, Dublanche et al 2006, Zhang et al 2012). Quantifying the effect of these inconsistent links can be challenging. The ‘teams’ conceptualization thus provides us with a way to quantify the extent of such inconsistencies through the lens of the impurity metric.By incorporating the notion of impurity, we substantially relaxed the strict definition of a team, enabling a broader application of this concept across diverse GRNs.
To investigate the functional implications of these structural properties, we simulated the dynamics of each network using the logical rules provided in the Cell Collective database, under an asynchronous update scheme (see Methods). For each network, we initialized 100 000 random initial conditions and recorded the final steady states. For each such state, we calculated a ‘team expression’ score, defined as the fraction of nodes within each team that were in the active (expression level 1) state. Similar to the cell fate decision networks earlier, the qualitative relationship between team strength, F1, and CCAB was maintained for Cell Collective GRNs as well (figure 2(B)). However, most of the networks had very low team strength, and in a low team strength regime, while F1 is consistently low, CCAB varied a lot, suggesting that team strength alone does not hold the differentiating power needed to predict the structure of the phenotypic landscape.
Across the Cell Collective GRNs, we observed an average network density of ∼15%, an average impurity of ∼30% (see figure 2(C)), and an average network size of 20 (figure S3). Despite using the biological rules for simulation as opposed to threshold-based rules as in figure X, we found that the correlation between the expression levels of the two teams—CCAB was strongly positive with impurity and negative with density (figures 2(D–i) and S2). Thus, impurity offers a better description of the phenotypic landscape than team strength. Networks with low impurity are closer in structure to two teams with mutual inhibition, leading to a strongly negative correlation between team scores. Whereas networks with a high impurity of (∼0.5) include the networks with mutually activating teams and therefore exhibit a strong positive correlation. Following our previous analysis of networks with teams (Hari et al 2022b), we calculated the frequency of ‘pure states’—F1, defined as the states with the team scores as 1,0, or 0,1. While the EMP networks showed a very high fraction of pure states (>75%, figure 1(C)), only three out of the 20 networks with teams in the Cell Collective database had high F1. F1 had a negative correlation with impurity (Spearman’s ρ = − 0.4, p-value < 0.05) and a positive correlation with density (Spearman’s ρ = 0.31, p-value < 0.05). (figure 2(D–ii)).
These findings suggest that a network’s structural characterization in terms of impurity and density can serve as a predictive indicator of its emergent phenotypic landscape. To probe this relationship further, we systematically generated artificial two-teamed networks (TTNs) spanning a range of impurity and density values and analyzed their dynamics using the same simulation framework. These results provide a unified framework to connect network architecture with phenotypic diversity, even in cases where traditional notions of well-separated teams break down.
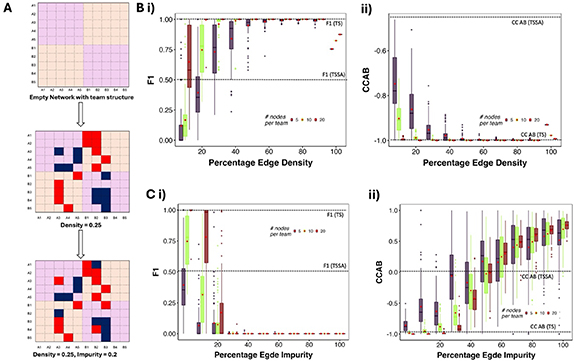
2.3. F1 and CC are strengthened with increasing density and team size, but weakened with increasing impurityWe generated artificial networks with two teams using the stochastic block model with constant probability for each community (Holland et al 1983), as described in figure 3(A). Briefly, we generated an adjacency matrix of size 2N × 2N filled with zeros, where N is the number of nodes in a team. Each element of the adjacency matrix represents an edge from the node corresponding to the row to the node corresponding to the column. We then fill the matrix with 1’s (in the submatrices corresponding to within team interactions, i.e. pink) and −1’s (in the submatrices corresponding to across team interactions, i.e. yellow) in randomly chosen positions in the matrix, such that the total number of non-zero elements Is 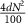 , where d is the required percentage edge density of the network. We then flip the signs of a fraction (
, where d is the required percentage edge density of the network. We then flip the signs of a fraction (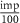 ) of the non-zero elements, where imp is the required percentage edge impurity. We generated an ensemble of the artificial TTNs, by varying 1) Team size (number of nodes in a team, changing as 5, 10 and 20), 2) Density (percentage of edges present in the network, ranges from 10% to 100%) and 3) Impurity (percentage of edges conflicting with the ‘team’ configuration, ranges from 0% to 100%), with 100 networks for each combination of parameters, totaling 33 000 networks. Note that we refer to density and impurity as ‘Core Edge Density’ and ‘Core Edge Impurity’, where core edges refer to edges within the TTN. We use this terminology to differentiate the properties of the core network from the peripheral/external network later in the embedding simulations done later described in figure 5. The range of team size was determined to capture the mean network size (20—captured by team size 10) seen in Cell Collective networks.
) of the non-zero elements, where imp is the required percentage edge impurity. We generated an ensemble of the artificial TTNs, by varying 1) Team size (number of nodes in a team, changing as 5, 10 and 20), 2) Density (percentage of edges present in the network, ranges from 10% to 100%) and 3) Impurity (percentage of edges conflicting with the ‘team’ configuration, ranges from 0% to 100%), with 100 networks for each combination of parameters, totaling 33 000 networks. Note that we refer to density and impurity as ‘Core Edge Density’ and ‘Core Edge Impurity’, where core edges refer to edges within the TTN. We use this terminology to differentiate the properties of the core network from the peripheral/external network later in the embedding simulations done later described in figure 5. The range of team size was determined to capture the mean network size (20—captured by team size 10) seen in Cell Collective networks.
Figure 3. F1 and CC AB as a function of the edge density and impurity. (A) Schematic representing the generation of two-teamed networks (TTNs). (B) Boxplots showing the comparison between distributions of (i) F1 and (ii) CCAB with variation in percentage edge density at zero impurity. the boxes are colored by the team size, with the corresponding legend displayed inside the plot. (C) Same as B, but variation against the percentage edge impurity at a constant density of 20%. For each boxplot, the corresponding mean values of the metric on the y axis (F1 or CCAB) are shown using red diamond-shaped points.
Download figure:
Standard image High-resolution imageWe simulate the TTNs along with the TS and TSSA networks using RACIPE (Huang et al 2018) and Boolean formalisms (Font-Clos et al 2018). RACIPE constructs and ODE system for a given network, then simulates it at a large ensemble of parameter sets to estimate the phenotypic landscape. The Boolean formalism used is a threshold-based formalism inspired by the Ising model of ferromagnetic systems. Among various threshold formalisms available, the formalism used here mimics the Boolean networks in literature best (Kadelka and Hari 2025). From both formalisms, we obtain steady-state frequency distributions as a measure of the phenotypic landscape. We evaluated the bimodality of the phenotypic landscape using F1—the total frequency of ‘pure’ phenotypes (where all the nodes of one team have high expression and all those of the other team have low expression, equivalent to 10 or 01 states in TS) and the expression patterns using CCAB—the strength of correlation between the ‘teams’ (see methods). We compared these against the calculations obtained from simulating TS and TSSA.
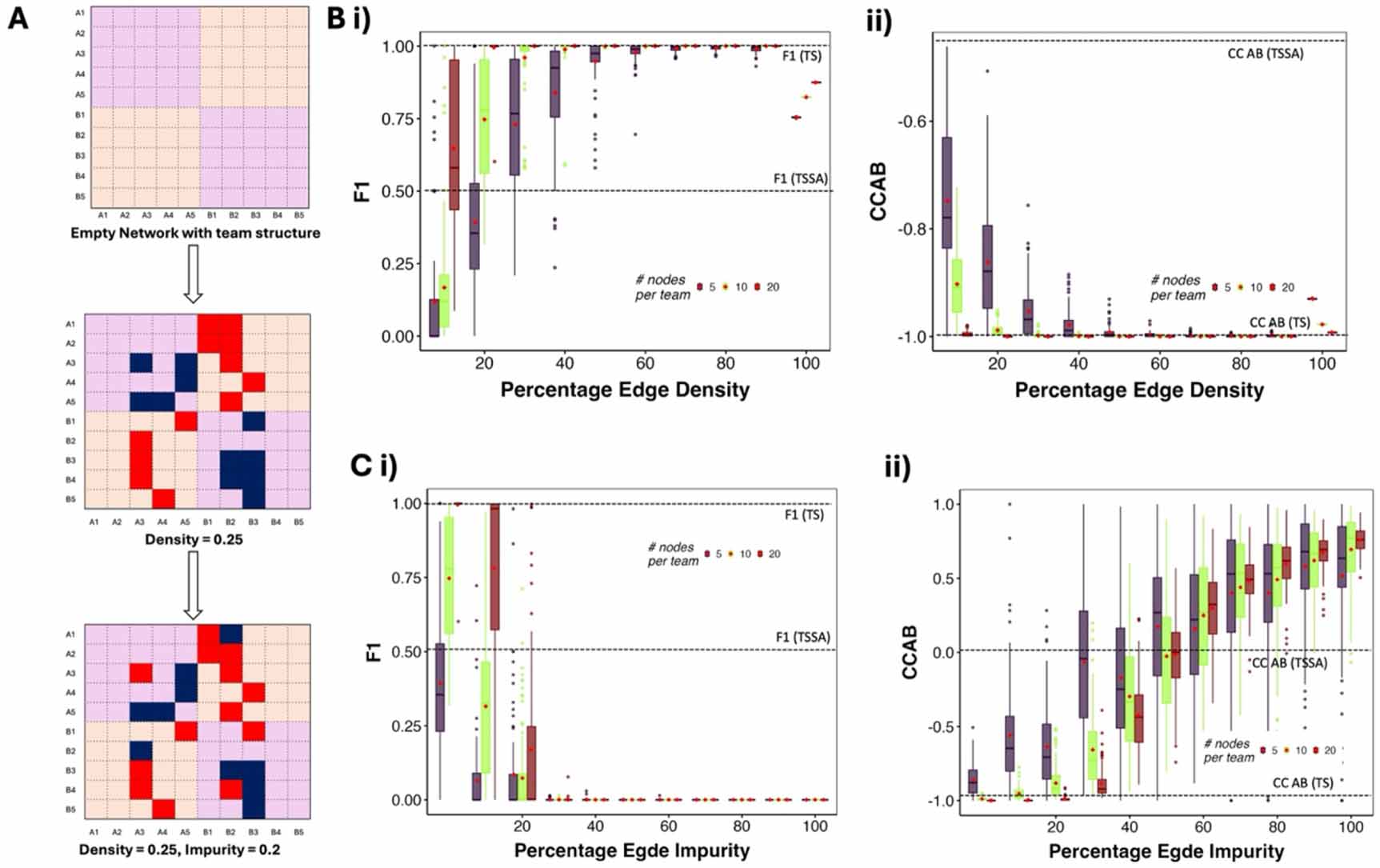
In the absence of any impurities (Hari et al 2025), the frequency of ‘pure’ (mutually exclusive) states (F1) increases with increasing density and increasing team size in both Boolean and continuous simulations (figures 3(B), S4(A) and (D), Hari et al 2025). While networks with 20 nodes per team saturated quickly, reaching F1 of 1 at 20% density, smaller networks required larger densities. The networks with 20 nodes showed a mean F1 of 0.75 at 20% density and saturated at 30% density. At 60% density, even the smaller networks reach an F1 of 1, the same as a TS. The F1 of TSSA, believed to be the core network of many cell-fate decision systems, is 0.5. We find that most networks of team size 10 cross that threshold at 20% density, the mean density of Cell Collective networks. Team size 5 networks required 40% density to cross the threshold F1 of 0.5. This complementary relationship between team size and density could be the reason for the negative correlation between team size and density found in the Cell Collective.
In Boolean simulations, the maximum value of F1 is 1, and in continuous simulations, it is around 0.85. In both cases, the maximum F1 of TTN networks is equal to or above the F1 values obtained from a TS (as shown by the horizontal lines). Interestingly, at 100% density in both Boolean and continuous simulations, F1 values decreased from their maximum to approximately 0.8. This observation is analogous to the increase in the frequency of hybrid states in TSSA as compared to TS (Hari et al 2022a, F1 in figure 3(B–i)). We hypothesize that up to 90% density, the activations within teams contribute towards making the teams work as single entities, beyond which the activation links within a team amount to adding a self-activation to the super-nodes, thereby increasing the occurrence of ‘hybrid’ phenotypes. To further understand this behavior, we removed all activation links from the TTN networks and found that F1 monotonically increased with density (figures S4(B) and (C)). It is important to note that some networks at low densities and small team sizes also achieve very high values of F1 (see the outlier dots at 10% and 20% densities). Biological networks also showed similar behavior at low density, where most of the networks have low F1, and a small fraction have higher F1.
The correlation coefficients of TTN networks were consitently negative. At lower densities (⩽20%), CC AB shows a higher variance. As the density increases, the variance decreases, with the mean approaching that of TS in both Boolean and continuous simulations (figures 3(B–ii) and S4(C)). Team size has a similar strengthening effect on correlation as density. Thus, TTN at any density can be simplified as a TS in the absence of impurities while studying the correlation between expression of teams/phenotypes. For the more nuanced studies involving the fraction of pure states in a population governed by these networks, tacitly simplifying TTN to TS or TSSA is not always valid. As biological networks have lower density, simplifying cell fate decision systems into simple TSs will lead to overestimation of pure states. Frustration of a steady state, a metric that measures the extent of inconsistency between the configuration of a steady state and the network topology, correlates negatively with the frequency of the steady state (Tripathi et al 2020, Hari et al 2022b). An activatory (inhibitory) edge is frustrated if the expression levels of the nodes corresponding to the edge do not have the same expression level (have the same expression level). Frustration of a state is defined as the fraction of such frustrated edges. In the absence of impurities, the ‘pure’ states (111 1100 000 or 000 0011 111 for a 10-node TTN) are not frustrated. However, as we introduce impurities in a TTN, the ‘pure states’ of the original TTNs become frustrated, with the value of frustration equal to the fraction of impure edges in the network. Given the previously observed negative correlation between frustration and steady state frequency (Hari et al 2022b), we predict the F1 metric to decrease as the number of impurities increases in the network. Indeed, we find that impurity has a negative effect on F1 at different team sizes (figures 3(C–i)). The mean F1 for all network sizes quickly reduced to zero as impurity increased to 30%. Even at 80% density, where networks of all sizes had an F1 of 1 at 0% impurity, the F1 decreased to 0% at 40% impurity (figure S5(A)), demonstrating the strong impact impurity has on the TTN phenotypic landscape. It is interesting to note that the average impurity of biological networks is 25%, and the density is around 20%. Thus, our results align with the low F1 values found in Cell Collective networks (figure 2(D ii)), despite the difference in simulation formalism. To better understand the decrease in F1, we studied additional metrics that relaxed the definition of F1 by including the frequency of states with deviation from pure states up to one and two nodes (figure S5(B)), thereby creating two relaxed F1 metrics. While the mean values of these relaxed F1 metrics are larger than those of the strict F1, we still see a sudden decrease in the mean values of these metrics from 20% to 40% impurity. A subset of networks do show higher values of the relaxed metrics as well as the strict F1, suggesting the existence of additional network features that can regulate the effect of impurity on F1. From 60% impurity onwards, both the relaxed F1 metrics have zero values as well.
CC AB provides a better understanding of the changes in the phenotypic landscape as compared to F1 with changing impurity. The mean correlation strength between the two teams reduces as impurities are introduced, nearing zero as the impurities reach 50%. With further increase in impurities, the correlation coefficient changes sign from negative to positive and starts increasing, reaching 1 at 100% impurity (figures 3(C–ii) and S5(C)). At 100% impurity, the TTN is effectively no longer a mutually inhibiting teams’ case, but a mutually activating one, leading to strongly positive CCAB. Correspondingly, we see the frequency of states with both teams ‘on’ or both teams ‘off’ increase with higher impurity (Figure S5D). Similar to F1, at 20% impurity, more than 75% of the networks show a stronger correlation than TSSA, but most networks have a weaker correlation than TS (−1). At 60% impurity, a significant fraction of the networks (>25%) still showed negative CC AB. While at 80% and 100% impurity, when the edges with each team are mostly inhibiting and those across teams are mostly activating, we still see a small number of networks showing negative CC AB.
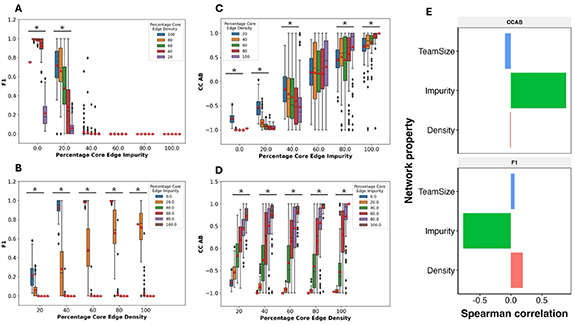
2.4. Impurities have a stronger effect on the phenotypic landscape than density and team sizeAfter analyzing the effect of density and impurity individually, we investigated the combined effect of different impurity and density values on F1 and CC of the TTN networks. At all densities, the networks with 60% or higher impurities show an F1 value of 0, indicating that high impurity can impair the TTN functioning irrespective of density. At 20% impurity, we observe a non-zero mean F1 for all densities as seen earlier (compare figure 4(A) with figure 3(C)). As density increases, we see an increase in the mean value of F1 for networks with 20% impurity, indicating that density can make the phenotypic output resilient to these impurities. Note that the magnitude of the increase in mean F1 is lower, from 80% to 100%, as compared to other consecutive density points. However, at 100% density, the mean F1 for 20% impurity networks is very similar as that at 0% impurity, while there is a strong decrease in mean F1 for other densities (figure 4(B)). This observation indicates that high density prioritizes negating the effects of impurity over adding self-activation to the network. At all other values of density, we see a clear decrease in mean F1 as impurity increases (figure 4(B)). The median values of F1 (horizontal dashes within the boxplots) follow the same trend as that of the mean. First, we take a closer look at the networks with 20% impurity in figure 4(B) (orange boxes). The range of F1 values is 0–1 for densities 40% to 100%. At 20 and 40% density, a large fraction of the networks have low F1 values. The span of the inter-quartile range (IQR) for F1 decreases from 40% to 100% density. However, the upper limit of the IQR increases from 40%–80% density, but goes back down at 100% density, reminiscent of the decrease observed in figure 3(B). The lower limit of the IQR consistently increases with an increase in the density. We interpret this behavior in the following manner. On average, an increase in density increases F1 while an increase in impurity reduces F1. Near the lower limit of the IQR, impurity dominates, while near the upper limit, density dominates, evident by the decrease in F1 at 100% density as compared to 80% density. However, other network features can impact this visible competition between impurity and density.
Figure 4. Varying the percentage of impure edges and density together. Comparison between the distributions of (A) F1 and (C) CC AB for TTNs of team size 5, having the same Percentage Core Edge Impurities but different Percentage Edge Densities. Comparison between the distributions of (B) F1 and (D) CC AB for TTS-5 networks, having the same percentage core edge density but different Percentage Edge Impurities. * indicates that the p-value of one-way ANOVA is <0.05. (E) Bar plot depicting the Pearson’s correlation coefficient of Network parameters, percentage core edge density, and impurity and Team Size with CCAB (top) and F1 (bottom).
Download figure:
Standard image High-resolution imageThe correlation coefficient reveals further interesting observations in this regard. As seen in figure 3(C), the correlation coefficient moves from value close to −1 to a value close to 1 as impurity increases, following a shift in the network structure from mutually inhibiting teams to mutually activating teams (figure 4(C)). As the density increases, the range of CC values over the same range of impurity values increases (figure 4(d)). We found similar dependence of F1 and CC AB on density and impurity for team size of 10 (figure S6). When density is varied at constant values of impurity, the direction of change in correlation coefficient depends on the impurity value (figure 3(B)). For impurity less than 50%, we see a decrease in the correlation coefficient (i.e. CC becomes more negative), whereas after 50%, CC increases, becoming more positive. Note that below 50% impurity, mutual inhibition of teams is dominant, while above 50%, mutual activation is dominant, possibly explaining the above-mentioned trend. Overall, we find that the increase in density amplifies the impact of the dominant behavior of the network in terms of the correlation coefficient. Thus, unlike in cases of no impurity (figure 3(C)), we see a clear effect of density on CC in the presence of impurities.
While F1 is a strict measure (figure S5), CC AB is more flexible. At 40% impurity, we do not see any networks with an F1 of 1, and a very low fraction of networks with non-zero F1. However, at 40% and 60% impurity values, we see networks with 1 and −1 correlation coefficients at all densities above 20%. While the changes in mean F1 and CC are captured well by the density and impurity of the TTN networks, the wide range of values suggests that predicting these metrics for individual networks would require additional understanding of the network topology.
To calculate the relative effect of density and impurity on F1 and CC, we calculated pairwise correlation between the density and the impurity vectors with F1 and CCAB (figure 4(E)) vectors. In other words, we measured the association of each parameter with F1 and CCAB as the other parameter(s) is varied over a wide range. As expected, impurity has a negative (positive) correlation with F1 (CC AB) while density has a positive (negative) correlation with these metrics correspondingly. We find that impurity has a stronger correlation with both F1 and CC as compared to density, suggesting that impurity has a stronger impact on both these metrics. Because F1 falls to zero after 40% impurity, we studied the networks with impurity 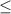 40% separately and found that impurity still has a stronger correlation with both F1 and CCAB than density does. At a constant impurity, the effect of density on CCAB is much lower (figure 3(C)) as compared to the effect of impurity at a constant density (figure 3(D)).
40% separately and found that impurity still has a stronger correlation with both F1 and CCAB than density does. At a constant impurity, the effect of density on CCAB is much lower (figure 3(C)) as compared to the effect of impurity at a constant density (figure 3(D)).
One factor often ignored in GRN-based models is the interaction of these networks with the rest of the signaling networks within which they are embedded. Indeed, biological networks have been argued to be modular (Serban 2020, Alcalá-Corona et al 2021, Kadelka et al 2023), but multiple phenotypic plasticity programs are interlinked, such as EMT, stemness, and drug resistance through a few intermediary molecules and pathways (Pasani et al 2020, Bangarh et al 2024). Thus, any network reduction strategy in terms of identifying teams of nodes should be capable of recapitulating the effect of such global interactions. We had previously shown that: (1) TS motif is resilient to perturbations from such ‘global’ interactions to a large extent (Harlapur et al 2022). (2) The correlation and PC1 variance of larger networks are significantly affected by the presence of a sub-network with teams (Hari et al 2025). These observations indicate that teams can provide resilience to global interactions similar to that seen in a TS. Thus, we investigated the effect of global interactions on the phenotypic outcome of TTN.
We embedded TTNs in a larger external network of 30 nodes, sampling external network parameters from the ranges mentioned in table 1 (see schematic in figure S8). We investigated three external network parameters that can potentially influence the dynamics of the core network: (1) mean connectivity, a measure similar to density that describes the number of connections each node can have within a network on average. (2) Average indegree to the core node, measured as the average number of connections from the external network to the nodes of the core network, and (3) Average outdegree of the core node, measured as the number of edges from the nodes of the core network that can affect the core network dynamics via feedback through the external network and the indegree to the core network. We first generated the core and external networks separately. The core network was generated using the stochastic block model as described earlier, varying the core edge density, core edge impurity, and team size as required for the particular experiments (figure S8(A)). For the external network, we generated a matrix of dimensions 30 × 30 filled with zeros, where each cell represents an edge from the corresponding node in the row to that in the column. We then randomly chose 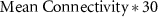 edges in the matrix and assigned 1 (activation) or −1 (inhibition) to these edges with equal probability (figure S8(B)). We then combined the core and external matrices as shown in figure S8(C). We then added
edges in the matrix and assigned 1 (activation) or −1 (inhibition) to these edges with equal probability (figure S8(B)). We then combined the core and external matrices as shown in figure S8(C). We then added 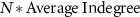 edges (randomly choosing activations and inhibitions with equal probability) to the top right rectangle that contains the edges from the external network to the core network and
edges (randomly choosing activations and inhibitions with equal probability) to the top right rectangle that contains the edges from the external network to the core network and 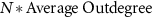 edges to the bottom left rectangle that contains the edges from the core network to the external network, where N is the number of nodes in the network (figures S8(D) and (E)). Note that the mean connectivity relates to the density of the external network as follows:
edges to the bottom left rectangle that contains the edges from the core network to the external network, where N is the number of nodes in the network (figures S8(D) and (E)). Note that the mean connectivity relates to the density of the external network as follows:
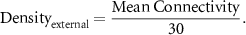
Table 1. Ranges for different parameters considered for network embedding.
Network parameterRangePercentage core edge densityFeasible densities > 20%Percentage core edge impurityFeasible impurities > 20%Mean connectivity of external networkMean connectivity ∈ [2,11]Average indegree of core nodeAverage indegree ∈ Average outdegree of core nodeAverage indegree ∈We refer to the mean connectivity instead of density for the external network so that it remains distinct from core edge density. The choice for the range of mean connectivity was inspired by the distributions of connectivity reported for an ensemble of biological networks (Kadelka et al 2024). We first wanted to test the effect of core network parameters on F1 of an embedded TTN, starting with the team size. We generated 100 such networks for each value of core team size (varying from 1 to 6) with 100% core edge density, 0% core edge impurity, and chose the external network parameters randomly from the ranges described in table 1. Apart from team size 2, TTN shows higher mean F1 values than the TS when embedded in a larger network (figure 5(A), compare team size 1 with higher team sizes). The mean F1 saturates at 1 for team sizes above 5, suggesting that larger TTN networks are more resilien
Comments (0)