
Remember me


Diabetes increases the risk of developing HF and arrhythmia. To study pro-arrhythmic remodelling, we performed Western blot analysis of ventricular tissue from patients with dilated cardiomyopathy and insulin-requiring diabetes (DiabDCM), comparing them to hearts from non-diabetic patients with dilated cardiomyopathy (DCM) and to non-failing (NF) hearts.
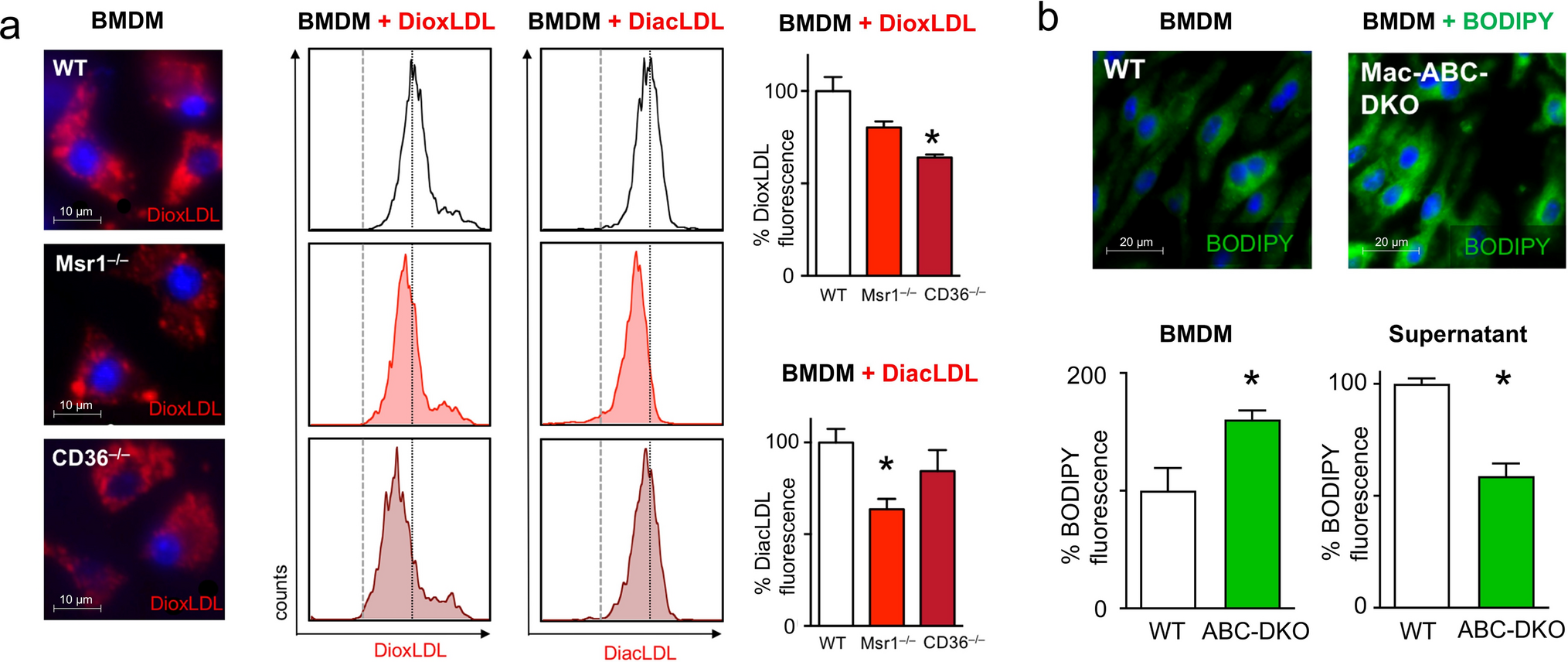
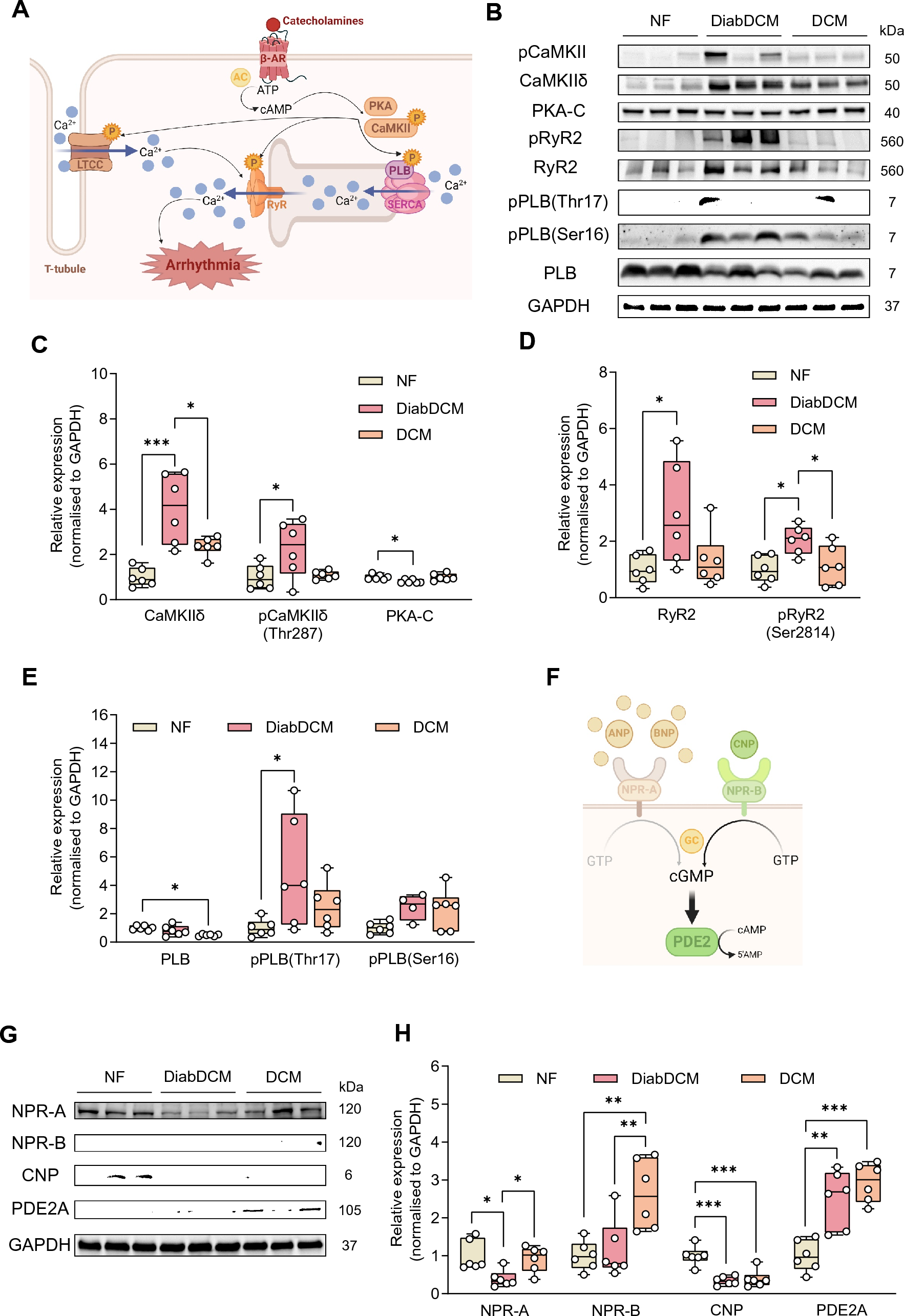
Sympathetic activation in response to cardiac dysfunction elevates intracellular cAMP levels, thereby activating cardiac kinases downstream of β-adrenoreceptors (β-AR), such as Ca2+/calmodulin-dependent kinase II (CaMKII) and protein kinase A (PKA) (Fig. 1A). Interestingly, expression and phosphorylation of CaMKII were markedly increased in DiabDCM compared to NF or DCM heart tissue, whereas the expression of the catalytic subunit of PKA (PKA-C) was reduced (Fig. 1B, C). One key target of CaMKII- and PKA-mediated phosphorylation is the ryanodine receptor (RyR2), which mediates Ca2+-induced Ca2+ release from the sarcoplasmic reticulum (SR) during systole. Hyperactivation has been demonstrated to contribute to arrhythmogenesis due to spontaneous Ca2+ releases during diastole (Fig. 1A). Notably, both RyR2 expression as well as its CaMKII-dependent phosphorylation at serine 2814 (pRyR2-Ser2814) were elevated in DiabDCM hearts compared to NF or DCM tissues, suggesting an enhanced activity especially in diabetic patients (Fig. 1B, D). During diastole, phospholamban (PLB) regulates the Ca2+ reuptake into the SR (Fig. 1A). In comparison with NF, PLB expression was reduced in DCM, but unchanged in tissues from DiabDCM. However, its CaMKII-mediated phosphorylation at threonine 17 (pPLB-Thr17) was increased in DiabDCM, but not in Hearts from DCM. The phosphorylation level at serine 16 (pPLB-Ser16, the PKA site) remained unchanged in both (Fig. 1E). These findings indicate pronounced pro-arrhythmogenic remodelling and pathophysiological hyperphosphorylation of Ca2+ cycling proteins via CaMKII in DCM patients with insulin-requiring diabetes.
Fig. 1
Pro-arrhythmogenic remodelling in patients with dilated cardiomyopathy with (DiabDCM) or without (DCM) insulin-requiring diabetes compared with non-failing controls (NF). A Illustration of proteins involved in β-adrenoreceptor (β-AR) signalling and pro-arrhythmogenic remodelling (created using BioRender.com). B Representative Western blots and quantification of the indicated protein expression and phosphorylation: C Ca2+/calmodulin-dependent kinase II (CaMKII) and its phosphorylation at threonine 287 (pCaMKII-Thr287), protein kinase A, catalytic subunit (PKA-C), D ryanodine receptor type 2 (RyR2) and its phosphorylation at serine 2814 (pRyR2-Ser2814), E phospholamban (PLB) and its phosphorylation at threonine 17 (pPLB-Thr17), and at serine 16 (pPLB-Ser16). F Schematic illustrating modulation of cAMP levels by natriuretic peptides via cGMP-dependent stimulation of phosphodiesterase 2 (PDE2) (created using BioRender.com). G Representative Western blots and quantification of indicated protein expression: H natriuretic peptide receptor A (NPR-A), natriuretic peptide receptor B (NPR-B), C-type natriuretic peptide (CNP), cGMP-dependent PDE2, N = 4–6 per group. Protein expressions were normalised to GAPDH, phosphorylation was normalised to total protein. Data are presented as box plots with whiskers showing minimum to maximum values, median and interquartile range. According to D’Agostino Pearson test, a normal distribution was assumed for all data. P values were determined by one-way ANOVA followed by Šídák’s multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001
By activating their receptors (NPR-A and NPR-B), natriuretic peptides increase intracellular cGMP levels, thereby indirectly stimulating PDE2 (Fig. 1F). Previously, we demonstrated anti-arrhythmic effects of PDE2 in healthy mice mediated via the CNP/NPR-B/cGMP axis [5]. Consequently, we examined the expression of NPRs, CNP and PDE2 in DiabDCM tissue (Fig. 1G). Compared with NF and DCM tissues, the protein expression of the ANP/BNP receptor NPR-A was markedly downregulated in DiabDCM hearts. In contrast, the expression of the CNP-receptor NPR-B was similar in hearts from DiabDCM and NF but enhanced in non-diabetic DCM patients. Furthermore, CNP expression was decreased in both patients with and without diabetes, whereas the expression of PDE2 was clearly upregulated in DCM and DiabDCM (Fig. 1H). Taken together, these results suggest a reduced ANP/BNP signalling and a CNP deficiency in DiabDCM hearts alongside elevated PDE2 abundance.
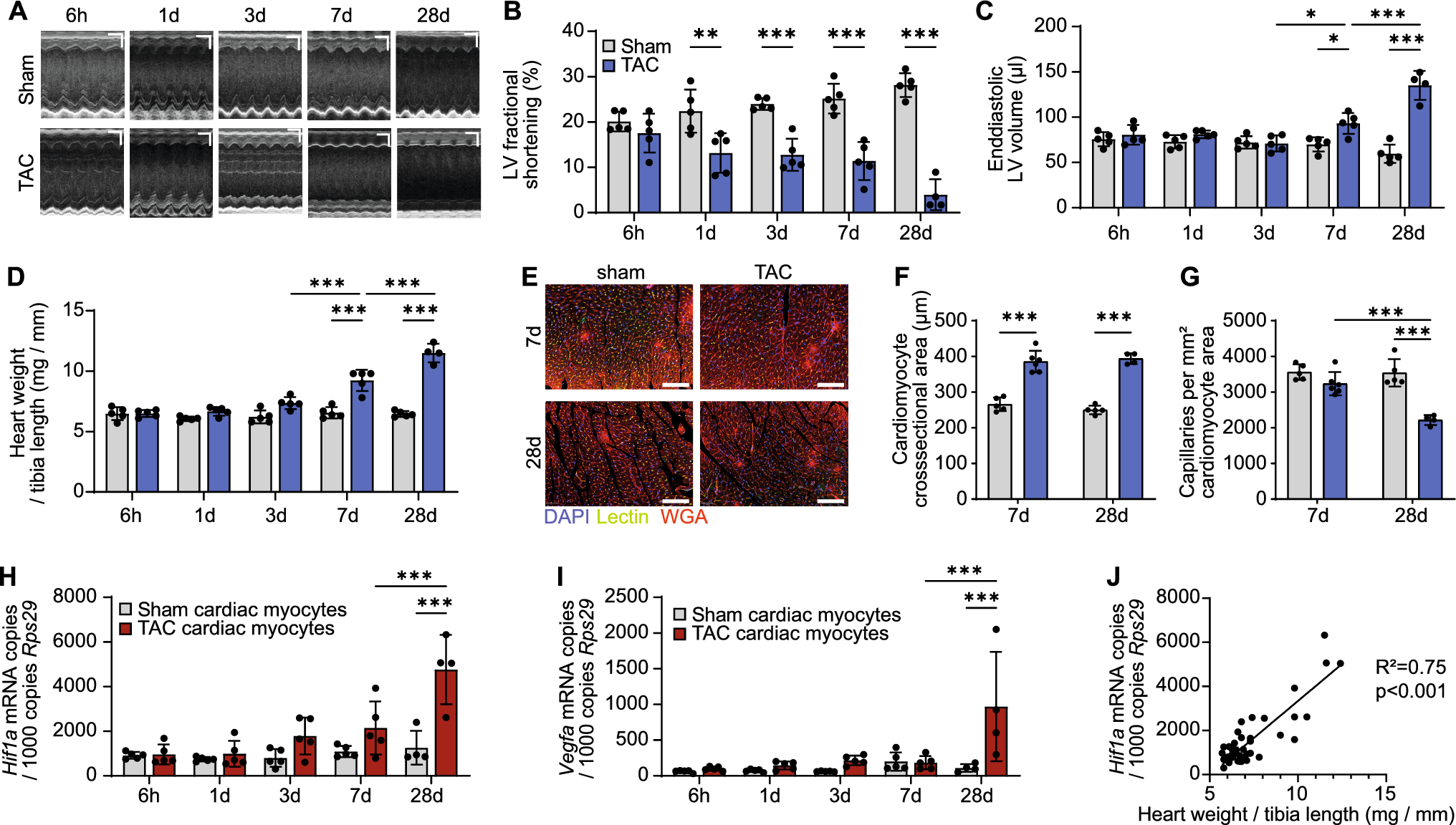
Failing hearts from STZ-treated mice show pro-arrhythmogenic cardiac remodelling resulting in enhanced arrhythmia susceptibilityTo investigate the cellular consequences of diabetes-induced pro-arrhythmic remodelling, wild-type mice were subjected to the well-established STZ T1DM model using five consecutive daily injections of STZ (50 µg/g i.p.). After 5 weeks, STZ-treated mice (STZ) exhibited elevated plasma glucose levels and HbA1c values compared to vehicle-treated controls (Con) (Supplementary Fig. S1A, B). STZ-induced hyperglycaemia was associated with impaired cardiac function as revealed by echocardiography (Supplementary Fig. S1C). Compared with controls, STZ treatment caused a mild reduction of left ventricular ejection fraction (LVEF) and fractional area shortening (FAS) resulting in decreased cardiac outputs (CO) (Supplementary Fig. S1D). Moreover, diastolic E/e’ ratios were clearly increased, indicating diastolic dysfunction (Supplementary Fig. S1E). Consistently, cardiac expression of the stress markers, atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP), was upregulated in hearts from diabetic mice (Supplementary Fig. S1F).
To validate the comparability of the STZ model with the human heart, the molecular composition of the proteins involved in the β-AR signalling pathway was investigated in ventricular cardiomyocytes from STZ-treated mice and compared to those from healthy controls (Con). Similar to the findings in DCM patients with diabetes, the expression and, notably, the phosphorylation levels of cAMP-related kinases and Ca2+-regulating proteins were altered in cardiomyocytes from diabetic mice (Fig. 2A), potentially contributing to impaired cardiac performance. Compared to Con, cardiomyocytes from diabetic mice displayed a reduced expression of PKA-C, indicating a reduced PKA activity. In contrast, CaMKII expression was unchanged. However, its phosphorylation at threonine 287 (pCaMKII-Thr287) was clearly higher, indicating increased activity (Fig. 2B). Whilst RyR2 expression levels were unaltered in cardiomyocytes from STZ-treated mice, this led to enhanced phosphorylation of RyR2 at the CaMKII site Ser2814 and was further reflected by elevated phosphorylation of PLB at the CaMKII site threonine 17 (pPLB-Thr17). In contrast, the PKA-dependent phosphorylation at serine 16 (pPLB-Ser16) was markedly reduced (Fig. 2C). Taken together, hearts from STZ-treated animals, like those of DiabDCM patients, exhibit altered protein expression within the β-adrenergic cAMP signalling pathway, characterised by an enhanced role of CaMKII and CaMKII-dependent phosphorylation.
Fig. 2
Compared with controls (Con), hearts and cardiomyocytes from mice with STZ-induced diabetes (STZ) exhibit pro-arrhythmogenic remodelling and increased arrhythmia susceptibility following ischaemia–reperfusion injury (I/R) or β-adrenergic stimulation. A Representative Western blots and quantification of indicated protein expression and phosphorylation in isolated ventricular cardiomyocytes from Con and STZ: B Ca2+/calmodulin-dependent kinase II (CaMKII) and its phosphorylation at threonine 287 (pCaMKII-Thr287), protein kinase A, catalytic subunit (PKA-C), C ryanodine receptor type 2 (RyR2) and its phosphorylation at serine 2814 (pRyR2-Ser2814), phospholamban (PLB) and its phosphorylation at threonine 17 (pPLB-Thr17), and serine 16 (pPLB-Ser16), D natriuretic peptide receptor B (NPR-B) and phosphodiesterase 2 (PDE2), N = 3–10 per group. Protein expressions were normalised to GAPDH, phosphorylation was normalised to total protein. E Representative ECG traces of ex vivo perfused Con and STZ hearts after I/R. F Total number of arrhythmic events and G mean heart rates (30 min after ex vivo I/R), N = 8 per group. H Representative Ca2+ spark (CaSp) recordings from Con and STZ cardiomyocytes under basal conditions (Ctrl), or following stimulation with Iso (10 nM) for 7 min and pacing at 1 Hz, 10 mV for 10 s. I Quantification of CaSp frequency (CaSpF) and SR Ca.2+ leak upon the respective conditions. n = number of cells / N = number of animals: Con: Ctrl (30/4), Iso (31/4); STZ: Ctrl (49/7), Iso (49/7). Data are presented as box plots with whiskers showing minimum to maximum values, median and interquartile range. According to D’Agostino Pearson test, a normal distribution was assumed for B, C: pRyR2-Ser2814, PLB, pPLB-Thr17, pPLB-Ser16, D and F, whereas a non-normal distribution was assumed for C: RyR2, G, and I. P values were determined by t test with (C: PLB) or without Welch’s correction (B, C: pRyR2-Ser2814, pPLB-Thr17, pPLB-Ser16, D, F), Mann–Whitney test (C: RyR2, G) or two-way ANOVA (I). *p < 0.05, **p < 0.01, ***p < 0.001
Diabetes is associated with an increased risk of AMI, ventricular arrhythmia and ultimately SCD [1]. To assess arrhythmia susceptibility in hearts from diabetic mice, the occurrence of arrhythmia was quantified following ischaemia induction by transient ligation of the left anterior descending artery (LAD) for 30 min followed by reperfusion (I/R) in ex vivo perfused diabetic hearts and compared to non-diabetic control hearts. Hearts from STZ-treated wild-type mice exhibited higher numbers of arrhythmic events after I/R than those from non-diabetic vehicle-treated controls (Con) (Fig. 2E). Notably, the overall number of arrhythmic events was markedly increased (Fig. 2F), whereas heart rates remained unchanged (Fig. 2G). The incidence of ventricular extrasystoles (VES), bigeminy and couplets also tended to be increased in STZ hearts (Supplementary Fig. S1G). At the cellular level, isolated cardiomyocytes from STZ-treated animals displayed a greater number of spontaneous Ca2+ releases (Ca2+ sparks, CaSp) from the SR in response to β-adrenergic stimulation with isoprenaline (Iso, 10 nM) compared to cells from healthy controls. Furthermore, this led to an increased total SR Ca2+ leak, reflecting CaSp amplitude, duration, width, and frequency, in cells from diabetic mice (Fig. 2H,I). Interestingly, the increased arrhythmia susceptibility observed in hearts and cardiomyocytes from diabetic mice was accompanied by elevated expression of NPR-B and PDE2 (Fig. 2D).
Pharmacological PDE2 activation by CNP and its analogue vosoritide reduces isoprenaline-induced pro-arrhythmogenic Ca2+ sparks in cardiomyocytes from diabetic micePreviously, we demonstrated that cGMP-dependent activation of PDE2 via CNP effectively suppresses pro-arrhythmic Ca2+ signals induced by β-adrenergic stimulation in cardiomyocytes from healthy mice [5]. We, therefore, investigated whether PDE2 stimulation also exerts anti-arrhythmic effects under diabetic conditions in cardiomyocytes from STZ-treated mice. Following the same protocol as previously published, we quantified spontaneous CaSp in isolated cardiomyocytes from diabetic wild-type (WT) mice (Supplementary Fig. 2A). Like in our experiments conducted in cardiomyocytes from healthy mice, β-adrenergic stimulation with Iso (10 nM) increased CaSp frequency (CaSpF) as well as the total SR Ca2+ leak. Importantly, CNP (1 µM) markedly reduced the Iso-induced CaSpF as well as the total Ca2+ leak, whereas concomitant PDE2 inhibition with the specific PDE2 inhibitor BAY 60–7550 (BAY, 100 nM) abolished the protective effects of CNP in cells from diabetic WT mice (Supplementary Fig. 2B). Moreover, the anti-arrhythmic effects of CNP were blunted in cardiomyocytes with cardiac-specific PDE2 knockout (KO) (Supplementary Fig. 2C, D). These findings indicate that, even under STZ-induced diabetic remodelling, CNP-dependent PDE2 activation mitigates pro-arrhythmic Ca2+ release.
However, as outlined above, the clinical application of CNP is limited by its short half-life. To overcome this, modified NPs were developed. The CNP analogue vosoritide (VO), which is approved for the therapy of achondroplasia, consists of the bioactive fragment CNP-53 with two additional Amino acids at the N-terminus, extending the plasma half-life to approximately 30 min (Fig. 3A) [6]. Given the anti-arrhythmogenic effects of CNP, we next investigated whether VO similarly reduces pro-arrhythmogenic CaSp. Indeed, under concomitant stimulation with Iso (10 nM), VO (1 µM) considerably suppressed the Iso-induced increase in CaSpF in cardiomyocytes from both control (Con) and STZ-treated (STZ) mice. Importantly, this effect was abolished by PDE2 inhibition with BAY (100 nM), indicating that VO exerts anti-arrhythmic effects via PDE2 activation (Fig. 3B, C). Similarly, VO reduced the total Iso-induced Ca2+ leak in cardiomyocytes from diabetic WT mice, whereas BAY co-treatment enhanced it (Supplementary Fig. S3A).
Fig. 3
cGMP-dependent PDE2 stimulation with vosoritide (VO) protects ventricular cardiomyocytes isolated from diabetic mice against pro-arrhythmogenic intracellular Ca.2+ sparks (CaSp). A Schematic structure showing the CNP analogue vosoritide (VO) and the bioactive CNP fragment CNP-53, created with Marvin for JavaScript. B Representative CaSp recordings in cells from wild-type mice without (Con) or with STZ-induced diabetes (STZ) under basal conditions (Ctrl), or following stimulation with Iso (10 nM), Iso + VO (1 µM), or Iso + VO + BAY 60–7550 (BAY, 100 nM) for 7 min and pacing at 1 Hz, 10 mV for 10 s. C Quantification of CaSp frequency (CaSpF) under the respective conditions, n = number of cells / N = number of animals: Con: Ctrl (30/4), Iso (33/4), Iso + VO (31/4), Iso + VO + BAY (35/4); STZ: Ctrl (50/7), Iso (50/7), Iso + VO (52/7), Iso + VO + BAY (49/7). D Representative CaSp recordings in cells from control animals (Con) and mice with STZ-induced diabetes (STZ) under basal conditions (Ctrl), or following stimulation with Iso (30 nM), Iso + VO (1 µM), or Iso + VO + BAY (100 nM) for 7 min and pacing at 1 Hz, 10 mV for 10 s. E Quantification of CaSpF under the respective conditions, n = number of cells / N = number of animals: Con: Ctrl (34/4), Iso (35/4), Iso + VO (37/4), Iso + VO + BAY (32/4); STZ: Ctrl (51/7), Iso (49/7), Iso + VO (48/7), Iso + VO + BAY (55/7). Data are presented as box plots with whiskers showing minimum to maximum values, median and interquartile range. According to D’Agostino Pearson test, all data were assumed to be non-normally distributed. p values were determined by Bonferroni test after a hierarchical model. *p < 0.05, **p < 0.01, ***p < 0.001
Cardiac dysfunction is associated with sympathetic overactivity, which enhances β-adrenergic stress in the heart and thereby promotes arrhythmogenesis [13]. To mimic this condition, we increased the Iso concentration to 30 nM in our experimental protocol and tested whether the anti-arrhythmogenic effect of VO (1 µM) was preserved under higher β-adrenergic stimulation. Interestingly, whilst the protective effect of VO was blunted in Con cardiomyocytes co-stimulated with Iso (30 nM), VO still mediated a relevant anti-arrhythmogenic effect in cardiomyocytes from STZ-treated mice (Cohen’s d = 1.096) (Fig. 3D, E, Supplementary Fig. S3B). Notably, this effect was abolished by co-incubation with BAY (100 nM) suggesting that the upregulation of PDE2 in hearts from STZ-treated mice amplifies the protective action of VO.
Vosoritide protects cardiomyocytes from diabetic mice against β-adrenergic stimulation-induced arrhythmogenic Ca2+ influxes and Ca2+ wavesCa2+ influx via L-type Ca2+ channels triggers a Ca2+-mediated Ca2+ release from the SR, thereby contributing to arrhythmogenesis [28]. To further study the potential anti-arrhythmic effects of VO, we analysed the effects of VO-mediated PDE2 activation on L-type Ca2+ current (ICa,L) density under β-adrenergic stimulation. Importantly, the Iso-induced increase in peak ICa,L density at 0 mV was clearly diminished by VO (Cohen’s d = 1.620), whereas co-application of the PDE2 inhibitor BAY reversed this effect, restoring Ca2+ influx in WT cardiomyocytes from diabetic mice (Fig. 4A, B).
Fig. 4
Vosoritide (VO) reduces pro-arrhythmogenic Ca2+ influx and spontaneous Ca2+ waves via PDE2 in ventricular cardiomyocytes isolated from diabetic mice. A Representative ICa,L recordings in cells from diabetic wild-type mice under basal conditions (Ctrl), or following stimulation with Iso (30 nM), Iso + VO (10 µM), or Iso + VO + BAY 60–7550 (BAY, 300 nM) for 10 min. B ICa,L current density measured at 0 mV under the respective conditions, n = number of cells / N = number of animals: Ctrl (13/10), Iso (17/10), Iso + VO (18/9), Iso + VO + BAY (14/7). C Representative traces of ventricular cardiomyocytes from diabetic wild-type mice loaded with Fura-2, under basal conditions (Ctrl), or following stimulation with Iso (30 nM), Iso + VO (10 μM), or Iso + VO + BAY (300 nM) for 10 min and subjected to arrhythmia provocation (pacing at 2 Hz, 10 mV for 30 s). D Quantification of spontaneous calcium waves (SCW) per cell, E Ca2+ transient amplitude at 2 Hz and F Ca.2+ transient decay at 2 Hz under the respective conditions, n = number of cells / N = number of animals: Ctrl (20/6), Iso (23/6), Iso + VO (23/6), Iso + VO + BAY (21/6). Data are presented as box plots with whiskers showing minimum to maximum values, median, and interquartile range. According to D’Agostino Pearson test, a normal distribution was assumed for B and E, whereas a non-normal distribution was assumed for D and F. p values were determined by Bonferroni test after a hierarchical model. *p < 0.05, **p < 0.01, ***p < 0.001
So far, we demonstrated that VO suppresses small Iso-induced Ca2+ releases via both L-type Ca2+ channels at the plasma membrane and RyR channels in the SR through PDE2 activation. However, the enhanced ICa,L as well as multiple CaSp can trigger large SR Ca2+ releases, resulting in arrhythmogenic Ca2+ waves and contraction. To test whether VO protects against such events, we assessed intracellular Ca2+ transients and the occurrence of spontaneous Ca2+ waves (SCW) after pacing at 2 Hz in cardiomyocytes from diabetic mice (Fig. 4C). Iso markedly increased the number of SCW in cardiomyocytes from diabetic animals, whereas VO substantially reduced Iso-induced SCW (Cohen’s d = 1.047), indicating a suppression of pro-arrhythmic contractions. This effect was abolished by simultaneous PDE2 inhibition with BAY (Fig. 4D). In cardiomyocytes from diabetic mice, the Ca2+ transient amplitude was not statistically significantly affected by Iso, VO or BAY (Fig. 4E). In contrast, Iso accelerated SR Ca2+ reuptake kinetics, whilst concomitant VO stimulation had no additional effect. PDE2 inhibition with BAY slightly slowed the Iso-induced time constant τ of the Ca2+ decay (Fig. 4F). Together, these results demonstrate that VO prevents arrhythmogenic Ca2+ waves in cardiomyocytes from diabetic mice, without impairing rhythmic Ca2+ cycling during β-adrenergic stimulation.
Genetic PDE2 deletion prevents cellular anti-arrhythmogenic effects of vosoritideTo validate that the anti-arrhythmogenic effects of VO are mediated via PDE2, pro-arrhythmic Ca2+ releases and currents in isolated cardiomyocytes were quantified in diabetic mice with cardiac-specific PDE2 deletion (PDE2 KO) subjected to STZ treatment. Consistent with the effects of pharmacological PDE2 inhibition, VO did not affect the Iso-induced increase in CaSpF or the SR Ca2+ leak in cells from diabetic PDE2 KO animals (Supplementary Fig. S4A, B). Similarly, patch-clamp experiments revealed that genetic PDE2 deletion prevented the VO-mediated reduction of ICa,L, displaying comparable current densities at 0 mV under stimulation with both, Iso alone or Iso plus VO (Supplementary Fig. S4C, D). VO also failed to decrease Iso-mediated pro-arrhythmogenic SCW in PDE2-deficient cardiomyocytes, whilst Ca2+ transient amplitudes remained unaffected, as in WT cells (Supplementary Fig. S4E). Together, these results indicate that VO protects against pro-arrhythmic Ca2+ signals in cardiomyocytes from diabetic mice via cGMP-dependent PDE2 activation, an effect lost upon genetic deletion or pharmacological inhibition of PDE2. This supports the critical role of the NPR-B/cGMP/PDE2/cAMP axis in limiting pro-arrhythmogenic Ca2+ releases in cardiomyocytes.
Vosoritide prevents arrhythmia generation ex vivo after ischaemia–reperfusion injuryAs a proof of concept for the potential anti-arrhythmic effect of VO, we perfused isolated hearts from diabetic WT mice and quantified arrhythmic events after I/R (Fig. 5A). In diabetic hearts, VO markedly reduced the occurrence of arrhythmic events (Cohen’s d = 2.340), whereas co-perfusion with the PDE2 inhibitor BAY abolished this protective effect (Fig. 5B). When differentiating amongst arrhythmia types, VO decreased the number of VES, bigeminy, triplets and couplets, whilst BAY increased their occurrence (Fig. 5C). These results highlight the anti-arrhythmic potential of vosoritide after AMI in hearts of diabetic individuals.
Fig. 5
Vosoritide (VO) protects hearts from diabetic mice against arrhythmia following ex vivo ischaemia/reperfusion injury (I/R) by stimulating PDE2 and reducing cAMP/CaMKII-dependent phosphorylation of Ca2+ cycling proteins at the cellular level. A Representative ECG traces, B total number of arrhythmic events, C number of ventricular extrasystoles (VES), tachycardia, bigeminy, couplets and triplets, in hearts from STZ-treated mice perfused ex vivo with Krebs–Henseleit buffer containing physiological catecholamine (CA) concentrations (10 nM norepinephrine, 3.5 nM epinephrine) with or without vosoritide (200 nm), or BAY 60-7550 (BAY, 300 nm) after I/R, N = 6 per group. D cAMP content in isolated ventricular cardiomyocytes quantified by direct cAMP ELISA following treatment with Iso (10 nM), Iso + VO (1 µM), or Iso + VO + BAY (100 nM) for 10 min, N = 6 per group. Relative cAMP concentrations were normalised to corresponding sample protein content and the Iso group. E Representative Western blots, and F quantification of Ca2+/calmodulin-dependent kinase II (CaMKII) phosphorylation at threonine 287 (pCaMKII-Thr287) and CaMKII-dependent phosphorylation of ryanodine receptor 2 at serine 2814 (pRyR2-Ser2814) and phospholamban at threonine 17 (pPLB-Thr17) in murine ventricular heart tissue following ex vivo Langendorff perfusion with Krebs–Henseleit buffer containing catecholamines (CA, 10 nM norepinephrine, 3.5 nM epinephrine), CA + VO (200 nM), or CA + VO + BAY (300 nM) for 1:15 h, N = 6 (CA, CA + VO + BAY), or N = 7 (CA + VO) per group. Protein phosphorylation was normalised to total protein. Data are presented as box plots with whiskers showing minimum to maximum values, median, and interquartile range. According to D’Agostino Pearson test, normal distribution was assumed for B, C: bigeminy, couplets, D, F: pCaMKII-Thr287, pPLB-Thr17, non-normal distribution was assumed for C: VES, tachycardia, triplets and F: pRyR2-Ser2814. p values were determined by Dunnett’s T3 multiple comparisons test after Brown–Forsythe test and Welch ANOVA (B, C: bigeminy, F: pPLB-Thr17), Šídák’s multiple comparisons test after one-way ANOVA (C: couplets, F: pCaMKII-Thr287), RM one-way ANOVA (D), or Dunn’s multiple comparisons test after Kruskal–Wallis test (C: VES, tachycardia, triplets, F: pRyR2-Ser2814). *p < 0.05, **p < 0.01
Vosoritide reduces catecholamine-induced cellular cAMP levels and phosphorylation of Ca2+ handling proteinsTo validate the proposed mechanism that VO reduces β-AR/cAMP-mediated arrhythmia development in diabetic cardiomyopathy via PDE2 activation, intracellular cAMP levels were measured in isolated cardiomyocytes from STZ-treated mice. ELISA assay revealed that VO clearly attenuated Iso-induced intracellular cAMP accumulation, whereas simultaneous PDE2 inhibition restored cAMP levels (Fig. 5D). The downstream effects of VO-mediated intracellular cAMP reduction on phosphorylation of CaMKII at threonine 287 (pCaMKII-Thr287), as well as its targets RyR2 at serine 2814 (pRyR2-Ser2814) and PLB at threonine 17 (pPLB-Thr17), which contribute to arrhythmogenesis, were quantified (Fig. 5E). Normalised to total protein expression levels, VO reduced CaMKII phosphorylation, indicating decreased kinase activity, whilst PDE2 inhibition reversed this effect. Consistently, Iso-induced phosphorylation of RyR2 and PLB at their CaMKII sites was diminished by VO, likely mediating reduced pro-arrhythmogenic SR Ca2+ releases. This effect was prevented by PDE2 inhibition, which restored RyR2 and PLB phosphorylation levels (Fig. 5F). These results provide mechanistic evidence that the anti-arrhythmic effects of VO are mediated by reduced intracellular cAMP levels and consequent suppression of CaMKII-dependent phosphorylation of Ca2+ handling proteins.
Vosoritide does not affect cardiac action potentials and cellular contractionNext, we assessed the impact of VO on electrophysiological and contractile properties of cardiomyocytes isolated from diabetic mice. In ex vivo perfused diabetic hearts, VO did not alter QT interval or heart rate compared to controls. (Fig. 6A, B). At the cellular level, action potentials (AP) were recorded in isolated cardiomyocytes from diabetic WT mice in the absence and presence of VO (Fig. 6C). VO did not affect key AP parameters, including resting membrane potential, maximal upstroke velocity, or AP duration at 50 and 90% repolarisation (Fig. 6D).
Fig. 6
Vosoritide (VO) does not affect heart rate or QT interval in ex vivo perfused hearts, nor action potential and contraction properties of ventricular cardiomyocytes isolated from diabetic mice. A Heart rate in diabetic hearts perfused ex vivo with Krebs–Henseleit buffer containing physiological catecholamine concentrations (10 nM norepinephrine, 3.5 nM epinephrine) without (Ctrl) or with VO (200 nM), N = 6 per group. B Representative ECG recordings and quantification of QT intervals of diabetic hearts perfused ex vivo with Krebs–Henseleit buffer containing physiological catecholamine concentrations (10 nM norepinephrine, 3.5 nM epinephrine) without (Ctrl) or with VO (200 nM), N = 6 per group. C Representative action potential recordings in cardiomyocytes kept in bath solution with or without VO (10 µM). D Baseline membrane potential, depolarisation velocity, and action potential duration at 50% (APD50) and 90% (APD90) repolarisation, n = number of cells / N = number of animals: Ctrl (7/4), VO (8/4). E Representative traces of sarcomere shortening under control conditions (Ctrl) and following stimulation with VO (1 µM), Iso (10 nM), or Iso + VO for 5 min and pacing at 1 Hz, 10 mV. F Sarcomere shortening amplitude normalised to baseline sarcomere length, contraction velocity and relaxation velocity in cardiomyocytes under the respective conditions, n = number of cells / N = number of animals: Ctrl (58/6), VO (63/6), Iso (56/6), Iso + VO (60/6). Data are presented as box plots with whiskers showing minimum to maximum values, median and interquartile range. According to D’Agostino Pearson test, a normal distribution was assumed for A, B, D, whereas a non-normal distribution was assumed for F. p values were determined by t test (A, B, D) or by Bonferroni test after a hierarchical model (F). ***p < 0.001
To evaluate contractile function, fractional shortening and contraction/relaxation velocities were measured in cardiomyocytes from STZ-treated WT under Iso, VO, or PDE2 inhibition (Fig. 6E). As expected, Iso enhanced the fractional cell shortening compared to controls. In contrast, VO did not affect basal fractional shortening or basal contraction and relaxation velocities, nor did it affect the Iso-induced enhancement of contraction parameters in cardiomyocytes from diabetic mice (Fig. 6F). These results indicate that VO exerts its anti-arrhythmic effects without altering baseline electrophysiology or contractile function in cardiomyocytes from diabetic animals.
The anti-arrhythmogenic effects of vosoritide persist in human iPSC-derived cardiomyocytesFinally, we tested whether VO exerts anti-arrhythmic effects in human cardiomyocytes. Therefore, hiPSC-CMs were cultured under high glucose conditions (22 mM glucose, HG) to mimic a diabetic environment [25], whilst hiPSC-CMs cultured in medium with standard glucose conditions (11 mM glucose, NG) served as control (Fig. 7A). Stimulation of the cells with Iso increased CaSp frequency and SR Ca2+ leak similarly in both NG and HG hiPSC-CMs. Remarkably, VO was potent in reducing Iso-induced spontaneous Ca2⁺ releases in HG but not in NG hiPSC-CMs, mirroring the observations in adult murine cardiomyocytes (Fig. 7B, Supplementary Fig. S5). Co-incubation with the PDE2 inhibitor BAY abolished the anti-arrhythmogenic effect of VO in hiPSC-CMs cultured under HG conditions (Fig. 7C, D). Multi-electrode array recordings further revealed that VO did not affect the beating rate-corrected field potential duration (FPDc), which reflects cardiac repolarisation, nor the conduction velocity or beating rate, in spontaneously beating HG hiPSC-CM cultures, regardless of β-adrenergic stimulation with Iso (Fig. 7D).
Fig. 7
Vosoritide (VO) reduces Ca2+ sparks (CaSp) in human iPSC-derived cardiomyocytes (hiPSC-CMs) cultured under high glucose (HG) conditions via activation of PDE2, without affecting cellular electrophysiological parameters. A Representative CaSp recordings of hiPSC-CMs cultured under normo (NG) or high (HG) glucose conditions under basal conditions (Ctrl), or following stimulation with Iso (100 nM), or Iso + VO (1 µM) for 5 min and pacing at 0.5 Hz, 10 mV. B Quantification of CaSp frequency under the respective conditions (CaSpF), n = number of cells / N = number of independent experiments: NG: Ctrl (22/4), Iso (28/4), Iso + VO (27/4); HG: Ctrl (17/3), Iso (33/4), Iso + VO (27/4). C Representative CaSp recordings of HG-hiPSC-CMs under basal conditions (Ctrl), or following stimulation with Iso (100 nM), Iso + VO (1 µM), or Iso + VO + BAY 60–7550 (BAY, 100 nM) for 5 min and pacing at 0.5 Hz, 10 mV. D Quantification of CaSpF and SR Ca.2+ leak under the respective conditions, n = number of cells / N = number of independent experiments: Ctrl (33/4), Iso (47/4), Iso + VO (38/4), Iso + VO + BAY (41/4). E Effects of VO (1 µM), Iso (100 nM) or Iso + VO on field potential duration corrected for beating frequency using Fridericia’s formula (FPDC,), conduction velocity and beating rate of HG-hiPSC-CMs. n = number of cells / N = number of independent experiments: Ctrl (11–12/2), VO (10/2), Iso (9–12/2), Iso + VO (7–8/2). Human iPSC-CMs were differentiated from iPSC lines created from a healthy donor and cultured in a medium containing a normal (Ng, 11 mM glucose) or high (HG, 22 mM glucose) concentration of glucose for 7 days. Data are presented as box plots with whiskers showing minimum to maximum values, median, and interquartile range. According to D’Agostino Pearson test, a normal distribution was assumed for E: conduction velocity, whereas a non-normal distribution was assumed for B, D, E: FPDC, beating rate. P values were determined by Bonferroni test after a hierarchical model. *p < 0.05, **p < 0.01, ***p < 0.001. F Schematic illustration of the CNP/vosoritide-activated cGMP/cAMP crosstalk in cardiomyocytes (created using BioRender.com)
Overall, VO reduced Iso-induced pro-arrhythmic Ca2⁺ signals—including CaSp, ICa,L amplitude and SCW—at the cellular level and decreased arrhythmia following I/R at the organ level in the STZ-induced diabetic mouse model. This anti-arrhythmic effect was recapitulated in hiPSC-CMs cultured under high glucose conditions (Supplementary Fig. S6). Mechanistically, VO-induced cGMP generation in cardiomyocytes exposed to hyperglycaemic conditions enhances PDE2 activity, lowers intracellular cAMP, reduces CaMKII activity and consequently diminishes spontaneous RyR2-mediated pro-arrhythmic Ca2⁺ releases (Fig. 7F).
Comments (0)