
Remember me
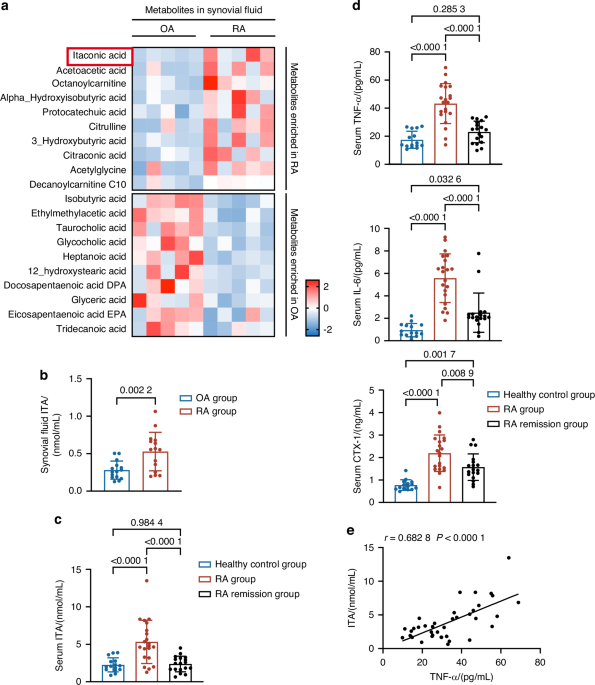
A total of 20 patients (OA = 13), who were admitted to the hospital for total hip arthroplasty (THA), were recruited in this research by the Department of Orthopedic Surgery, the Sixth People’s Hospital of Shanghai Jiao Tong University (Shanghai, China). Control human samples were obtained from individuals with femoral neck fractures with no history of arthritic ailment (n = 7). The clinical features of individuals with patients are given in Table S1. Every individual gave informed agreement to utilize their clinical data for scientific research. The samples were kept in tissue storage solution (Miltenyi Biotec) and shipped immediately after operation. This investigation was authorized by the Ethics Committee of the Sixth People’s Hospital of Shanghai Jiao Tong University. The approval number were 2023-KY-033(K) and 2024-KY-270(K).
Cartilages were excised from the hip femoral condyles, and synovial tissue was obtained from the hip joint. These specimens were kept at −80 °C. Synovial tissue samples from OA patients were divided into three sections (~5 × 5 × 3 mm3 for each section) and were treated with 2 mL of whole medium with or without EVs from ADSCs for 72 h in 12-well culture plates. Following incubation, synovial specimens were regularly immunohistochemistry stained. 5-μm-thick slices were treated overnight at 4 °C with primary antibodies specific for CD206 (1:4 000, Abcam, ab252921), iNOS (1:100, Abcam, ab115819). The portion was stained with an HRP detection system (Servicebio).
Animal modelOA rats modelMost induced OA models are done in males mice or rats, and the direct comparison of OA outcome measures or interventions are mainly based on male animals.77 Males animals treated with induced OA model may be more progressive, compared to the females animals.78 Male wild-type (WT) Sprague-Dawley (SD) rats, 6-week-old, were obtained from SLAC Laboratory Animal Co. Ltd. (Shanghai, China). Rats were housed at Tongji University Animal Unit under normal conditions (22 ± 2 °C ambient temperature, 50% ± 5% humidity and 12-h light cycle). When the rats were 8 weeks old, they were subjected to destabilization of the medial meniscus (DMM) and ACLT operations in the right knee joints to produce mechanical instability-related OA, as previously mentioned.79 Specifically, the anterior cruciate ligament on the right knee was transected surgically, and meanwhile the medial meniscus was destabilized by surgically dividing the medial meniscotibial ligament. For the sham operation, the right knee joint cavity was exposed after the incision of the cutaneous and muscular planes, and then closed with suture.
For the experiments in Fig. S1A, all rats undergoing DMM + ACLT operations or sham operations were arbitrarily divided into two groups: (1) sham group and (2) OA group. After 8 or 16 weeks of OA surgery, anesthesia of the rats was kept with isoflurane at each time point, the blood samples was taken in ethylenediaminetetraacetic acid tubes (BD) and the specimens were submitted to pathological investigation.
For the experiments in Fig. 1d, all rats undergoing DMM + ACLT operations or sham operations were randomly allocated into four distinct categories: (1) sham + PBS group; (2) sham + PBS-EVsM1 group; (3) OA + PBS group; (4) OA + PBS-EVsM1 group. Three weeks after the surgery, rats received several injections of 50 μL PBS or 50 μL PBS-EVsM1 (1 × 1010 particles/mL) intra-articularly throughout the next 6 weeks (twice a week). After 10 weeks of surgery, the rats’ anesthesia was kept up with isoflurane, and the specimens were submitted to pathological examination.
For the experiments in Fig. 2e, all rats undergoing DMM + ACLT operations or sham operations were arbitrarily allocated into five distinct categories: (1) sham group; (2) OA + PBS group; (3) OA + PBS-EVsM1 group; (4) OA + PBS-EVsM1/antagomiR-155-5p group; (5) OA + PBS-EVsM1/antagomiR-NC group. Rats were pre-injected with 50 μL antagomiR-NC or antagomiR-155-5p (5 nmol) for 8 weeks (two times a week), 1 week following an operation, and then given numerous intra-articular injections of 50 μL PBS or 50 μL EVsM1 (1 × 1010 particles/mL) for 6 weeks (two times a week). Following 10 weeks of operation, the rats’ anesthesia was kept up with isoflurane, and the specimens were submitted to pathological evaluation.
For the experiments in Fig. 6d, all rats undergoing DMM + ACLT operations or sham operations were arbitrarily allocated into five distinct categories: (1) sham group; (2) OA + PBS group; (3) OA + PBS-EVsADSCs group; (4) OA + PBS-EVsADSCs-antagomiR-155-5p group; (5) OA + PBS-MAP-EVsADSCs-antagomiR-NC group. Four weeks after the surgery, the rats received several intra-articular injections of 50 μL PBS or 50 μL EVsADSCs (1 × 1010 particles/mL) for 6 weeks (one time a week). After 12 weeks of operation, the rats’ anesthesia was kept up with isoflurane, and the specimens were sent for pathological investigation.
The Ethical Committee of Laboratory Animals Research Center, Tongji University, authorized the animal studies listed above. The authorization number is TJAA07622701. The National Institutes of Health’s Guidelines for the Care and Use of Laboratory Animals were followed for all animal-related experiments.
OA mice modelThe C57BL/6J-miR155em1(flox)Cya mice (miR-155flox/+) were acquired from Cyagen Biosciences Inc (Suzhou, China). The primers utilized for verifying the flox gene in mice are Forward (5′-TGGAATAAGTCACAAGGACAGTGA-3′) and Reverse (5′-CCAAGGGTTGAGAGGAGGA ATTTA-3′). The LysMCre mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA; No. 005680). Then, miR-155flox/flox mice were mated with LysMCre mice to produce myeloid lineage-specific miR-155 knockout mice (miR-155fl/fl; LysMCre). The C57BL/6J-Col2a1Cre mice were acquired from Cyagen Biosciences Inc (Suzhou, China, No. C001474). Then, miR-155flox/flox mice were mated with Col2a1Cre mice to produce chondrocytes-specific miR-155 knockout mice (miR-155fl/fl; Col2a1Cre). Regular genotyping of tail DNA was conducted as per the recommendations. In the Animal Unit of Tongji University, all mice were kept on a 12-h light cycle. The DMM or sham operations of mice were conducted on the right knee joints as described above.79 The Ethical Committee of Laboratory Animals Research Center, Tongji University, authorized the animal research described above. The authorized number is TJAA07622103. All animal treatments were conducted in conformity with the Guidelines for the Care and Use of Laboratory Animals of the National Institutes of Health.
For the experiments in Fig. S7K, all LysMCre; miR-155 fl/fl (CKO) mice (male and female) undergoing DMM operations were arbitrarily allocated into three distinct categories: (1) OA + PBS group; (2) OA + CM group; (3) OA + CM + AgomiR-155-5p group. Six weeks after the surgery, the mice received several intra-articular injections of 10 μL PBS or 10 μL CM or AgomiR-155-5p (1 nmol) for 6 weeks (twice time a week). After 13 weeks of operation, the mice’ anesthesia was kept up with isoflurane, and the specimens were sent for pathological investigation.
CellsThe primary macrophages derived from bone marrow (BMDMs) were collected and cultivated, as mentioned earlier.80 The cervical dislocation was used to euthanize SD rats, and the tibia and femur were both removed. The bone marrow was washed out with a 1 mL syringe and cold phosphate-buffered saline (PBS) with 1% penicillin-streptomycin (P/S). After filtering through a 70-μm strainer, 1 mL of red blood cell lysis solution was added to eliminate red blood cells (RBC) (Solarbio, China, R1010). After waiting 10 min, cells were centrifugated for 10 min at 450 × g, 4 °C, and the supernatant was discarded; 10 mL PBS was put into the tube and mixed. Following centrifugation, cells were rinsed two time and then were blocked with in PBS with rat Fc Block (BD, 550271) at 4 °C for 20 min. After rinsing with PBS, cells were reconstituted in the FACS buffer containing the following antibodies: PerCP/Cyanine5.5 anti-rat CD45 antibody (1 μg/mL, Biolegend, 202220), PE/Cyanine7 anti-rat CD11b/c antibody (2.5 μg/mL, Biolegend, 201818), for 30 min at at 4 °C, and then sorted with BD FACS Flow cytometry. The sorted BMDMs were resuspended in Iscove’s modified Dulbecco’s medium (Gibco, USA) comprising 10% fetal bovine serum (FBS), 1% P/S, and 20 ng/mL M-CSF (R&D System, USA). Non-adherent macrophage precursors were cultivated after 1 day, and the medium was substituted every 3 days. On the 7th day, the mature BMDMs were defined as M0 BMDMs. To promote M1-like polarization, M0 BMDMs were incubated with Lipopolysaccharide (LPS, 100 ng/mL, serotype O55:B5, Enzo), IFN-γ (20 ng/mL) for 48 h before being stimulated with 5 mmol/L ATP for a further 60 min to activate the inflammasome. Flow cytometry was utilized to determine polarization. Moreover, BMDMs-derived EVs were isolated from the supernatant for additional studies.
The isolation of primary articular chondrocytes was performed according to the study.81 Cartilage samples of newborn SD rats or C57 mice by first rinsing them in sterile PBS containing P/S, following by slicing. The cartilage was then enzymatically digested to recover cells using high-glucose Dulbecco’s modified Eagle medium (DMEM) (Gibco, USA) supplemented with 1% P/S and 0.2% type II collagenase (Gibco, 17101-015). The cell suspension was passed through a 70 μm cell strainer, and the obtained cells were centrifuged at 400 × g for 5 min. The resulting pellets, composed of primary chondrocytes, were reconstituted in DMEM/F12 medium (Gibco, USA) supplemented with 1% P/S, 10% FBS and 1% glutamine. Every other day, fresh medium was replaced. Cells from passage three were utilized. The isolation of primary synoviocytes from 8-week old SD rats was similar to the procedure of the previous research.32
Adipose tissue-derived MSCs (ADSCs) were isolated following previously published protocols.82 Subcutaneous adipose tissue was taken from healthy and younger donors via liposuction. The study protocols and ethical guidelines were approved by the People’s Liberation Army No. 85 Hospital in Shanghai, P.R. China (review serial number NO.2013/18).83 A written authorization was obtained from each donor participant. All procedures were conducted following the Helsinki Declaration and standard protocols. Adipose tissue weighing 500 mg was sliced into 1-mm3 fragments and subjected to a 45-min digestion with 0.1% collagenase I (Gibco, USA) after rinsing with PBS. Following digestion, an equal volume of complete culture medium (DMEM-F12 culture medium with 10% FBS, 1% P/S) was added, and the mixture was centrifuged at 1 000 r/min for 10 min. Cells were cultured in a complete medium containing 10 ng/mL bFGF (Stem Cell, USA) at a density of 1 × 106/mL. Following that, cells were resuspended in PBS and passed through a 40 μm cell strainer. Adherent cells were passaged at a 1:3–1:4 ratio upon reach 80%–90% confluence. ADSCs up to passage 7 were used for experiments. The Human MSC examination Kit (BD Biosciences) and flow cytometry (FACSCalibur, BD, NJ, USA) were employed to characterize ADSCs surface markers. Data was analyzed using Flow Jo V10 software.
All cell lines employed in this investigation were taken from the American Type Culture Collection (ATCC). THP-1 human monocyte cells were cultured in RPMI 1640 (VivaCell Biosciences, C3010-0500), comprising 10% FBS and 1% P/S. The cells were differentiated into macrophage-like cells treated with PMA (100 ng/mL) for 48 h, and were defined as M0-like THP-1 cells. Then, the medium was substituted with an EVs-depleted serum, and the cells were maintained for a 48-h rest period. To induce M1-like polarization, M0-like THP-1 cells were pretreated with LPS (500 ng/mL, serotype O55:B5, Enzo), IFN-γ (20 ng/mL) for 48 h and then treated with 5 mmol/L ATP for an additional 60 min to stimulate the inflammasome. Flow cytometry was utilized to determine polarization. Additionally, THP-1-derived EVs were isolated from the supernatant for further investigation.
HEK293T cells were kept in DMEM supplemented with 10% FBS and 1% P/S.
Normal human chondrocytes C28/I2 cells, were grown in DMEM supplemented with 10% FBS and 1% P/S.
All cells were kept in an environment comprising 5% CO2 at a temperature of 37 °C.
Plasmids constructionThe packaging plasmids: pCMV-dR8.2 (Addgene plasmid #8455) and pCMV-VSV-G (Addgene plasmid #8454) were gifts from Bob Weinberg.84 The original pLVX-shRNA2-ZsGreen1 (No. 632179) was purchased from Clontech, and ZsGreen1 was replaced with puromycin N-acetyltransferase to obtain pLVX-shRNA2-Puro. The scrambled and GSK-3β or FMR1-targeted shRNAs were cloned into pLVX-shRNA2-ZsGreen1 or pLVX-shRNA2-Puro. The original pLVX-IRES-ZsGreen1 (No. 632187) was purchased from Clontech, and ZsGreen1 was replaced with puromycin N-acetyltransferase to obtain pLVX-IRES-Puro. The FMR1 coding region was inserted into pLVX-IRES-Puro. The construction of miR-155-5p sponge was produced according to previous researches.85,86 Four identical complementary miRNA antisense binding sites (AATTACGATTAGCACTATCCCCAA) and some short linkers were designed according to the previous study.85 The sponges’ 5’ and 3’ ends included overhangs compatible with EcolI and BamHI restriction endonucleases. The plasmid pLVX-IRES-Puro was digested and connected with the above target sequences. The relative primer sequences are shown in Table S2.
For the production of engineering EVsADSCs, the original plasmid pEGFP-C1-RVG-Lamp2b presented by Prof. Matthew J. A. Wood’s lab from the University of Oxford45 was cloned into plasmids encoding Lamp2b, MAP-Lamp2b and MAP-EGFP-Lamp2b respectively, according to the previous report.44
Lentiviral production and transductionThe production of lentivirus was designed according to standard protocols. The vectors including: pLVX-shRNA2-Puro, pLVX-shRNA2-ZsGreen1, pLVX-shGSK3β-Puro, pLVX-shFMR1-Puro and pLVX-IRES-Puro were respectively transfected into HEK293T cells with packaging plasmids using Lipo293TM (Beyotime, C0521). The supernatants of the transfected HEK293T cells including the lentivirus were collected 2 days and 3 days post transfection and filtered with a 0.45 μm filter. For the lentiviral infection of Thp-1 cells, spinoculation (1 000 × g for 2 h, at 37 °C, 10 ng/mL polybrene) was used. Then Thp-1 cells were plated into 6-well plates, supplemented with 4 mL fresh medium and cultured for an additional 48 h. For the lentiviral infection of C28/I2 cells, the cells were cultured with the medium of the transfected HEK293T cells for nearly 48–72 h at 37 °C.
Extracellular vesicles and conditioned medium isolation and characterizationTotal EVs from BMDMs, THP-1 cells, or ADSCs-derived cell supernatant were isolated using the differential ultracentrifuge method.
For the extraction of the EVs from BMDMs, M0-like BMDMs, and M1-like BMDMs, the cell supernatants were rinsed two times with PBS and then grown in DMEM culture medium added with 10% EVs-depleted serum (bovine EVs were depleted using overnight centrifugation at 100 000 × g) for 48 h. Then, the cell supernatant was obtained.
Extracellular vesiclesTo extract the EVs from THP-1 cells and M0/M1-like THP-1 cells, the cell supernatants were rinsed two times with PBS, then seeded in RPMI 1640 medium added with 10% EVs-depleted serum for 48 h. Following that, the cell supernatant was gathered.
For the extraction of EVsADSCs, ADSCs which reached 80%–90% confluence during the second passage, were grown in DMEM-F12 culture medium added with 10% EVs-depleted serum for 24 h. Subsequently, the cell supernatant was obtained.
The aforementioned cell supernatant underwent sequential centrifugation steps at 300 × g for 20 min, 3 000 × g for 20 min, and 10 000 × g for 30 min to remove dead cells, cell debris and apoptotic cells. Following filtration through a 0.22 μm filter, the supernatant was transferred to ultracentrifuge tubes (Beckman, 355618) and centrifuged at 100 000 × g for 2 h to isolate EVs. Each pellet was reconstituted in 100 μL PBS, with all procedures conducted at 4 °C. The purified EVs were either used immediately or stored at −80 °C.
The extraction of plasma EVs was purified using the previous method.42 The preparation of blood samples was taken within 2 h at 4 °C. First of all, the blood samples were centrifuged for 10 min at 1 690 × g to obtain plasma, then the plasma was centrifuged for 30 min at 10 000 × g to obtain platelet-free plasma. Following filtration through a 0.22 μm filter, the supernatant was transferred to ultracentrifuge tubes (Beckman, 355618) and centrifuged at 100 000 × g for 2 h to isolate plasma EVs. Each pellet was reconstituted in 100 μL PBS, with all procedures conducted at 4 °C. The purified plasma EVs were either used immediately or stored at −80 °C.
The particle size distribution, purity and amount of EVs were evaluated using Nanoparticle Tracking Analysis (NTA) (NanoSight 300, Malvern Instruments Ltd, UK). Specimens were diluted to the required concentration in 1 mL of PBS before being loaded into the sample chamber. Laser light was used to track and illuminated particles undergoing Brownian motion. Each specimen underwent multiple measurements, with procedural records maintained. The scattering intensity, size distribution and particle concentration were examined using the Stokes-Einstein equation. The ultrastructure of EVs was further examined using transmission electron microscopic (TEM), and images were acquired with Hitachi HT7800 electron microscope (HT-7800, Hitachi, Japan).
For co-culture investigations of chondrocytes and BMDMs-EVs, the EVs recovered from the BMDMs culture supernatant were dyed with DiI fluorescent dye via the DiI fluorescent cell linker kit (Beyotime, C1991S), followed by rinsing in PBS and ultracentrifugation (100 000 × g for 70 min) at 4 °C. Lastly, DiI-labeled EVs were reconstituted in PBS for the following investigation with chondrocytes. Moreover, to detect the absorption of BMDMs-EVs by the damaged articular cartilage, the EVs obtained from the BMDMs culture supernatant were dyed with DiO fluorescent stain by means of the DiO fluorescent cell linker kit (Beyotime, C1993S), accompanied by rinsing in PBS and ultracentrifuged (100 000 × g for 70 min) at 4 °C. At last, DiO-labeled EVs were reconstituted in PBS for the next investigation on OA model in animals.
Conditioned mediumThe BM-derived monocytes of femurs from LysMCre; miR-155fl/fl (CKO) mice were separated, and the monocytes carried the markers of CD45+ and CD11b+ were sorted using flow cytometry (Fig. S7B). Then, the sorted BM-derived monocytes were induced to M1-like polarization, the conditioned medium were collected and depleted EVs using the differential ultracentrifuge method. The EV-depleted conditioned medium (from about 107 cell) was prepared into lyophilized powder, then was reconstituted in 100 μL PBS.
Flow cytometry and fluorescently stimulated cell sortingMacrophages were characterized by the expression of markers CD45+, CD11b+, Gr1+, F4/80+. Tibias and femurs from C57BL/6 mice were processed according to the methods described in the “Cell culture” section to isolate monocytes derived from blood. Following the breakdown of red blood cells, BM cells were inhibited for 10 min at 4 °C using PBS and mouse Fc Block (BD, 553141). After washing with PBS, cells were resuspended in FACS buffer containing the following antibodies: PerCP/Cyanine5.5 anti-mouse CD45 antibody (2.5 μg/mL, Biolegend, 103132), PE/Cyanine7 anti-mouse/human CD11b antibody (2.5 μg/mL, Biolegend, 101216), BV421 anti-mouse Ly-6G/Ly-6C antibody (2.5 μg/mL, BD, 562709), Alexa Fluor 647 anti-mouse F4/80 antibody (1 μg/mL, Biolegend, 123122), PE anti-mouse CD86 (1 μg/mL, Biolegend, 105007) for 30 min at at 4 °C. Flow cytometry was conducted using a CytoFLEX S, Beckman Coulter. The analysis of the data was done with FlowJo (v.7.6.5).
Immunofluorescence of cellsChondrocytes were stained following a standard protocol. They underwent three brief washes in cool PBS for 5 min each. They were then fixed in either methanol or 4% paraformaldehyde for 20 min, and blocked with a Blocking Buffer for 60 min at room temperature. After that, the chondrocytes were incubated overnight at 4 °C with the proper primary antibodies, accompanied by repeated washes in PBS for 5 min each. After incubation with the corresponding fluorochrome-conjugated secondary antibody diluted in antibody dilution buffer for 1 h, chondrocytes were kept in the dark and rewashed in PBS for 5 min each. These specimens were stained with DAPI (Sigma, 32670), and washed three times for 5 min each in PBS. Representative photos were captured using a laser scanning confocal microscope (Leica, TSC SP8). The primary antibodies used were mouse anti-COL2A1 (1:1 000, Invitrogen, MA5-12789), mouse anti-MMP13 (1:1 000, Invitrogen, MA5-14238). The Alexa Fluor 488 goat anti-mouse IgG secondary antibody (1:1 000, Invitrogen, A32723) was used as for secondary reactions. Four random fields from each group were selected and utilized for statistical examination.
Western blotThe chondrocyte and cartilage specimens were treated with the radioimmunoprecipitation assay (RIPA) lysis solution (Epizyme, PC101, China) that contained a protease suppressor cocktail (Epizyme, GRF101, China) and a phosphatase blocker cocktail (Epizyme, GRF102). For the rat plasma samples, EVs derived from plasma were purified by differential centrifugation. Then, the obtained EVs pellet was reconstituted in RIPA lysate buffer. The amount of protein was estimated via a BCA protein testing kit (Takara, T9300A), and the specimens were immediately boiled for 10 min with the addition of a loading buffer. An equal amount of protein extracts (20 μg) were put on a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel for electrophoresis and transferred to PVDF membranes. The PVDF membrane was then treated successively with both primary and secondary antibodies. Lastly, the increased chemiluminescence (BI, 20-500-120) was utilized to react with secondary antibodies and the photos were taken. The COL2A1-antibody (1:1 000, Abcam, ab188570), MMP13-antibody (1:1 000, Invitrogen, MA5-14238), Sox9-antibody (1:1 000, Abcam, ab185966), Beclin1-antibody (1:1 000, CST, 3495), LC3A/B-antibody (1:1 000, CST, 12741), ATG3-antibody (1:1 000, CST, 3415), ATG7-antibody (1:1 000, CST, 8558), LAMP1-antibody(1:1 000, Abcam, ab62562), GSK-3β-antibody (1: 1000, CST, 12456), p-GSK-3β-antibody (1:1 000, CST, 5558), p70S6-antibody (1:1000, CST, 9202), P-p70S6-antibody (1:1 000, CST, 9234), S6-antibody (1:1 000, CST, 2217), P-S6-antibody (1:1 000, CST, 4858), FMRP-antibody (1:1 000, CST, 4317), GAPDH-antibody (1:1 000, CST, 5174), β-actin-antibody (1:1 000, CST, 8457), anti-rabbit IgG, HRP-linked Antibody (CST, 7074), anti-mouse IgG, HRP-linked Antibody (CST, 7076) were utilized as the antibodies.
RNA extraction, reverse transcription, quantitative real-time (RT)-polymerase chain reaction (PCR)Total RNA was extracted from tissues or cultured cells and exosomes using Trizol reagent (Invitrogen, 15596018/10296028). For mRNA qRT-PCR, the first-strand cDNA was synthesized with HiScript® III 1st Strand cDNA Synthesis Kit (Vazyme, R312-02, China). The relative expression of mRNA was normalized to that of GAPDH (mRNA control) using the 2−ΔΔCt method. For miRNA qRT-PCR, miRNA was converted to cDNA using the specific stem-loop primer with HiScript® III 1st Strand cDNA Synthesis Kit (Vazyme, R312-02, China). The qRT-PCR assay was performed on a light cycler (Roche, Basel, Switzerland) using ChamQTM Universal SYBR® qPCR Master Mix Kit (Vazyme, Q711-03). The relative levels of miRNA were calculated by the 2−ΔΔCt method with U6 small nuclear RNA (U6 snRNA; miRNA control in cells and tissues), Caenorhabditis elegans miR-39-3p (cel-miR-39-3p; miRNA spike control in exosomes and different tissues), according to the the previous study.87 Primer sequences are shown in Table S2.
Sequencing of EVs miRNA derived from M0 BMDMs and M1 BMDMs in rats modelTotal RNAs of EVsBMDMs were isolated as previously described and used for miRNA sequencing. The Agilent Rat miRNA Microarray, Release 21.0 (8*15 K, Design ID: 070154) investigation and data assessment were performed by OE Biotechnology Co., Ltd. (Shanghai, China). The microarray has 758 probes for mature miRNAs of rats.
The NanoDrop ND-2000 was used to quantify total RNA (Thermo Scientific), while Agilent Bioanalyzer 2100 (Agilent Technologies) was used to evaluate RNA integrity. The specimen labeling, microarray hybridization, and rinsing were conducted with the manufacturer’s standard methods. RNA was dephosphorylated and denatured, followed by labeling with Cyanine-3-CTP. The labeled RNAs were purified and subsequently hybridized onto a microarray. The arrays were scanned using the Agilent Scanner G2505C (Agilent Technologies) after thorough cleaning.
Feature Extraction software (version 10.7.1.1, Agilent Technologies) was used to acquire raw data. Genespring software (version 14.8, Agilent Technologies) was employed for the basic assessment. First, the raw data was processed using the quantile technique. Probes with every specimen in one group tagged “Detected” were selected for further data processing. Differentially appeared miRNAs were subsequently recognized based on fold change, and the P value was calculated via the t-test. The criteria for increased or decreased regulated miRNAs was a fold change of at least ≥2.0 and P value of less than ≤0.05.
Tissues RNA sequencingCartilage samples from the control group (OA + PBS) and EVsM1 treated group (OA + EVsM1) rats were collected (n = 3/group). The entire RNA was then isolated, as previously reported.43 The RNA libraries were sequenced by LC Bio Technology CO., Ltd (Hangzhou, China) by means of the illumina NovaseqTM 6000 platform. The bioinformatics examination, comprising genes differentially expressed (DEGs), KEGG and GSEA enrichment, was performed by means of the OmicStudio instrument at https://www.omicstudio.cn/tool. The heatmap was produced using R (https://www.r-project.org/) on the OmicStudio platform (https://www.omicstudio.cn/tool).
Transmission electron microscopy (TEM) examination for animal cartilagesAfter the decalcification of the tissues, TEM analysis was conducted. Cartilage samples were incubated in 2.5% glutaraldehyde for at least 4 h at ambient temperature, followed by four rinses of 15 min with 0.1 mol/L phosphate buffer. Subsequently, samples were dehydrated using increasing concentrations of acetone, post-fixed with 1% Osmium tetroxide for 1–2 h at normal temperature, and inserted in Epon 812 resin. The materials were then sectioned into 70 nm thick slices and imaged using the JEOL JEM-1230 electron microscope.
Histological analysisAfter fixation in 4% paraformaldehyde for 24 h, the entire knee joints were decalcified at pH 7.4 using 0.5 mol/L EDTA, and subsequently inserted in paraffin. Slices of 5 μm thickness were cut for staining with Safranin O/Fast Green (SO & FG) (Solarbio, G1371) and hematoxylin and eosin (H&E) (Thermo Fisher, 7211 & 7111). The OARSI scoring system was used for cartilage destruction in SO & FG-stained areas in the medial joints, including medial femoral condyles and the medial tibial plateau. To grade OA severity of mice, 0–6 subjective grading system was performed according to the previous methods.88 To assess OA severity of rats, the score of combined grade (0–6 grade) and stage (0–4 stage) were recommended based on the previous reports.89,90 The evaluation of synovial membranes features- enlargement of the synovial lining cell layer, density of the resident cell, and inflammatory infiltrate- was conducted using H&E-stained slides. Changes were assessed using a four-point scale: none (score: 0), slight (score: 1), moderate (score: 2), and severe (score: 3).29 Two blinded and independent observers evaluated each parameter, and the average score was employed for statistical analysis.
Immunohistochemistry and immunofluorescence examinationFor immunohistochemistry staining, the 5-μm-thick slices were treated overnight at 4 °C with primary antibodies appropriate for MMP13 (1:500, Proteintech, 18615-1-AP), COL2A1 (1:200, Servicebio, GB11021), GSK-3β (1:1 000, CST, 12456), CD206 (0.1 μg/mL, Abcam, ab64693), iNOS (1:1 000, Abcam, ab283655). The slices were stained with an HRP detection system (Servicebio), and hematoxylin was employed to counterstain them.
For immunofluorescence staining, the portions were inhibited with a blocking buffer [1 × PBS (BI, 02-024-1ACS)/5% normal serum (Jackson lab, 005-000-121, USA)/0.3% Triton™X-100 (Sangon Biotech, A110694, Shanghai, China)] for 60 min at ambient temperature. Then, the specimens were kept overnight at 4 °C with primary antibodies appropriate for COL2A1 (1:1 000, Invitrogen, MA5-12789), mouse anti-MMP13 (1:1 000, Invitrogen, MA5-14238), GSK-3β (1:1 000, CST, 12456), F4/80 (1:100, Santa Cruz, sc-377009), iNOS (1:1 000, Abcam, ab210823), CD206 (1:1 000, Abcam, ab64693). After washing, the slices were incubated with secondary antibodies for 1 h at room temperature. The Alexa Fluor 488 goat anti-mouse IgG secondary antibody (1:1 000, Invitrogen, A32723) and Alexa Fluor 594 goat anti-rabbit IgG secondary antibody (1:1 000, Invitrogen, A32754) were used. The representative photos were taken via a laser scanning confocal microscope (Leica, TSC SP8). For each joint, the proportion of positive cells was calculated in 3–5 fields, and the median percentage was typical for each mouse. The quantitative analysis of IF staining was conducted in a double-blinded way.
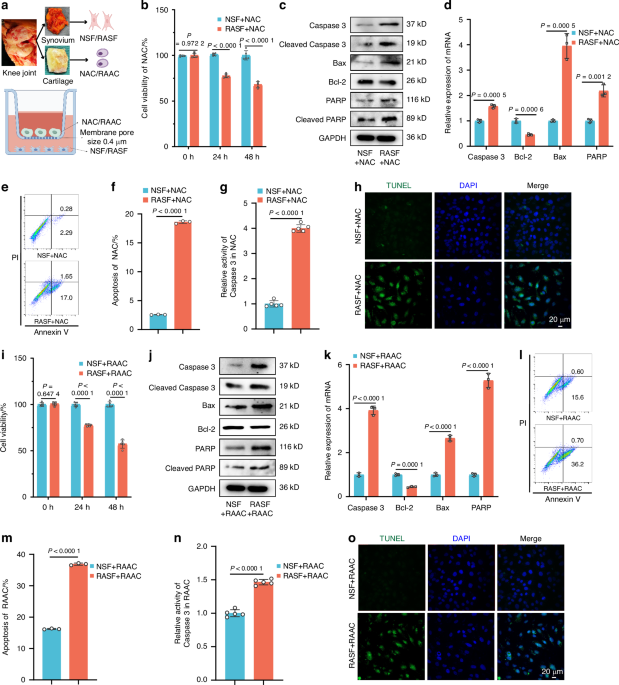
The co-culture assayThe THP-1 cells were resuspended at a density of 1 × 106 cells/mL well (2 mL) and seeded in the lower chambers (6-well migration chambers, 0.4 μm pore membrane, 83.3930.041, SARSTEDT, Germany), then treated into M1-like THP-1 cells with the previous method. The upper chambers were seeded with ADSCs of 1 × 105 cells/well in 6-well plates. After 72 h co-culture, the supernatant of the lower cells was used to isolate EVs using the differential ultracentrifuge method. Then, the EVs were analyzed for miRNA levels with qRT-PCR detection.
Autophagic flux assessmentThe mRFP-GFP-LC3 adenoviral vector (HB-AP2100001) was obtained from HANBIO. This vector exploits the pH difference between neutral autophagosome and acidic autolysosome to generate fluorescence, with red or red/green (yellow) fluorescence facilitating the tracking of autophagic flux. C28/I2 cells were seeded on coverslips in 24-well plates and treated with co-culture with CMM1/CMM0 or EVsM1-THP1/CMM0-THP1 or miR-155KD-EVsM0/M1/miR-NCKD-EVsM0/M1 or FRMPKD-EVsM0/M1/SCRKD-EVsM0/M1 or FRMPOE-EVsM0/M1/VectorOE-EVsM0/M1 for 48 h, up to the need of per experimental requirements to study autophagic flux in the cells. Subsequently, the mRFP-GFP-LC3 adenovirus was applied to the cells for 24 h. Following this, cells on coverslips were rinsed with cool PBS, fixed in 4% PFA, and dyed with DAPI for immunostaining of the nuclear. Images were captured using a laser scanning confocal microscope (Leica, TSC SP8). At least four images of mRFP-GFP-LC3-transfected cells were analyzed for each condition using a fluorescence microscope, and the percentage of transfected cells displaying mRFP-GFP-LC3 puncta was quantified to assess autophagosomes or autolysosomes formation.
Assay using a dual-Luciferase reporterThe onlined bioinformatics tool miRDB was employed to identify target genes and potential binding sites for miR-155-5p and GSK-3β. Wild-type (WT GSK-3β) and mutant GSK-3β dual-luciferase reporter vectors (MUT GSK-3β) were separately constructed and co-transfected into C28/I2 cells along with miR-155-5p mimic and the negative controls. Reporter assays were conducted following the supplier direction for the dual luciferase activity detection kit (Promega, E1910, USA), using a miRNA control as a negative control. Renilla luciferase activity was normalized to firefly luciferase activity and presented as a percentage relative to the control. The OD absorbance were measured using microplate reader (SpectraMax M5, Molecular Devices, SanJose, USA).
Elisa analysisTo quantify the levels of interleukin-10 (IL-10), interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α) and interleukin1β (IL-1β) in the rat serum, IL-10, IL-6 and IL-1β in the supernatants from clinical synovial tissue samples co-cultured with EVsADSCs, ELISA analysis with ELISA kits (Elabscience, Wuhan, China) were used following manufacturer recommendations. IL-10, IL-6, TNF-α, and IL-1β concentrations were calculated in pg/mL.
Statistical analysisThe number of samples for every experiment was established based on our previous experience. Different investigators supervised each of the experiments. All in vitro studies were replicated at least three times separately, and all in vivo tests were carried out on at least six physiologically independent animals from each group. All attempts to replicate the experiments were effective. GraphPad Prism 8.0 was employed for all statistical examinations (GraphPad Software Inc., La Jolla, CA, USA). Two-tailed Student’s t test (normal distribution) and Mann-Whitney U test (non-normal distribution) were used for comparisons between the two groups. One-way ANOVA & Tukey HSD post hoc test (normal distribution), Kruskal-Wallis Test & Dunn’s test (non-normal distribution) or Two-way ANOVA & Bonferroni test were used for multiple comparisons. All OARSI scoring were analyzed using non-parametric tests. Data are reported as the mean ± standard error (SEM), and P < 0.05 is considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.000 1, ns not significant.
Ethics approval and consent to participateAll animal experiments in the current study were approved by the ethics and technological committees of Tongji University. The approval number was TJAA07622701, TJAA07622103.
The clinical OA samples was obtained by total hip joint replacement surgeries. The clinical control samples was obtained from the individuals with femoral neck fracture with no history of arthritic diseases. All patients provided informed consent to use their clinical information for scientific research. This study was approved by the Ethics Committee of the Sixth People’s Hospital of Shanghai Jiao Tong University. The approval number was 2023-KY-033(K) and 2024-KY-270(K). The subcutaneous adipose tissue was obtained by liposuction from young healthy donors. The methods and guidelines were approved by the People’s Liberation Army No. 85 Hospital, Shanghai, P.R. China (review serial number NO.2013/18). All donors provided written informed permission.
Comments (0)