Mice and treatment
We purchased 3-month-old C57BL/6J (WT, Stock: 000664) mice from Jackson Laboratory (Bar Harbor, ME, USA). The Siglec-15 conditional knockout strain (C57BL/6 background) was obtained from the Mutant Mouse Regional Resource Center (MMRRC) and crossed with LysM-Cre mice from Jackson Laboratory to generate Siglec15ΔLysM mice (whole-body or myeloid-specific knockout), as previously described. Wild-type littermates from Siglec-15 heterozygotes were bred under the same conditions as the knockout mice for controls. Dmp1-Cre and RANKLfl/fl mice were purchased from Jackson Laboratory. Dmp1-Cre heterozygotes were crossed with RANKLfl/fl mice, and offspring were intercrossed to generate WT, Dmp1-Cre, RANKLfl/fl, and RANKLΔDmp1 (conditional Rankl deletion in DMP1+ cells) genotypes. Mice were euthanized at 4 or 11 weeks (10–12 per group) using isoflurane overdose for time-course studies. For sialidase injections, 4-week-old mice were anesthetized with ketamine (100 mg/kg, intraperitoneally) and xylazine (10 mg/kg, intraperitoneally). SialEXO 23 α2-3 specific sialidase (5 units, Genovis Inc.) was preincubated in 20 mmol/L Tris (pH 7.5, 37 °C, 1 h) and injected intrafemorally. All protocols were approved by the Army Medical University’s Animal Care and Use Committee.
RNA sequencing
Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Germany). Paired-end libraries were prepared with the TruSeq™ RNA Sample Preparation Kit (Illumina, USA) following the manufacturer’s protocol. Poly-A mRNA was purified using poly-T oligo-attached magnetic beads, fragmented at 94 °C for 8 min, and converted to cDNA through reverse transcription. Second-strand cDNA synthesis was performed using DNA Polymerase I and RNase H. The cDNA fragments were end-repaired, A-tailed, and ligated to adapters. After purification and PCR enrichment, libraries were quantified with a Qubit® 2.0 Fluorometer (Life Technologies, USA) and validated with an Agilent 2100 Bioanalyzer (Agilent Technologies, USA). Clusters were generated by cBot at 10 pmol/L concentration, and sequencing was conducted on the Illumina NovaSeq 6000 platform.
ATAC-seq and data analysis
ATAC-seq was performed on approximately 50 000 cells per sample to assess chromatin accessibility. Nuclei were isolated and subjected to tagmentation using Tn5 transposase, followed by PCR amplification to enrich for adapter-ligated fragments. Libraries were sequenced on an Illumina platform, and reads were quality-checked, trimmed, and aligned to the reference genome using Bowtie2. Peaks were called using MACS2, with differential accessibility analyzed via DiffBind. Peaks were annotated to nearby genes with ChIPseeker, and functional enrichment was assessed using GREAT. Data visualization and motif analysis were conducted using R packages and HOMER, respectively.
Chip-Seq
ChIP-seq experiments were conducted using the BeyoChIP™ Chromatin Immunoprecipitation Assay Kit with Protein A/G Magnetic Beads (P2080S, Beyotime). Sequencing libraries were prepared from 2–10 ng of purified ChIP DNA using the NEBNext Ultra DNA Library Prep Kit for Illumina (NEB) following the manufacturer’s protocol. The completed libraries were quantified using a Qubit fluorometer, Agilent TapeStation 2200, and RT-qPCR with the Kapa Biosystems library quantification kit. Uniquely indexed libraries were pooled in equimolar amounts and sequenced on the Illumina NextSeq500 platform with single-end 75 bp reads at the Dana-Farber Cancer Institute Molecular Biology Core Facilities. The sequencing data have been deposited in the GEO database under accession number GSE151481.
μCT analysis
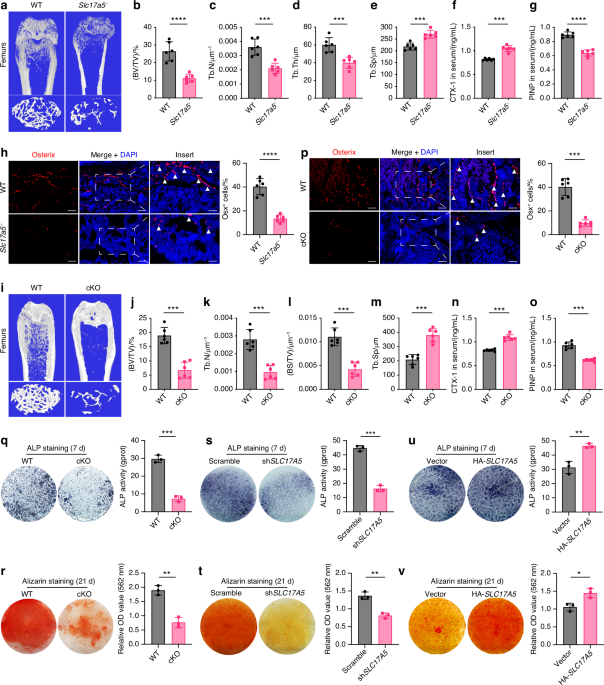
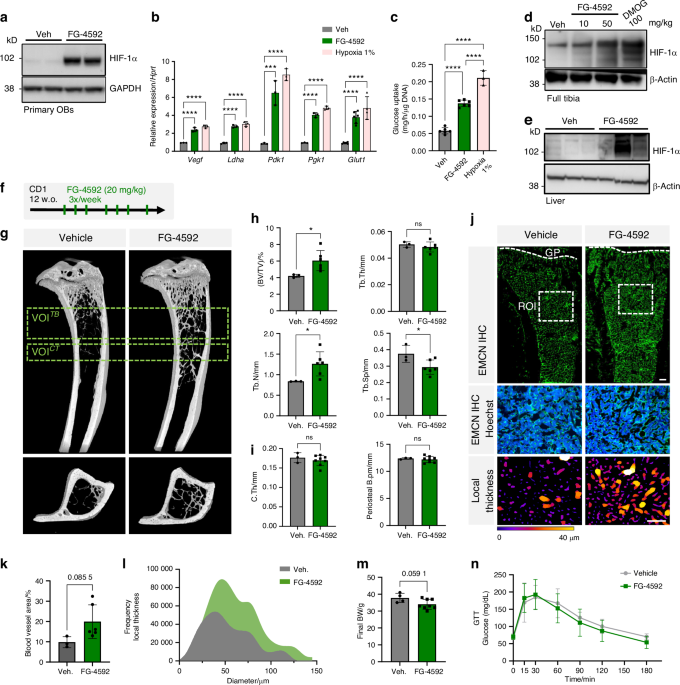
Mice were euthanized using isoflurane overdose, followed by perfusion with phosphate-buffered saline (PBS) for 5 min and 10% buffered formalin for another 5 min via the left ventricle. Femurs were dissected and fixed overnight in 70% ethanol, then analyzed using a Bruker μCT Skyscan 1172 system (Kontich, Belgium) at a 10.0 μm isotropic voxel size. Scanning was conducted at 60 kV, 166 μA, and 1 700 ms exposure. 3D reconstructions of the region of interest, including the DBM, were performed using NRecon software (Kontich, Belgium). 3D and 2D analyses were done with CT Analyser (Ver. 1.15.4.0, Kontich, Belgium), evaluating parameters such as Tb.Con, Tb.Sp, BV/TV, Tb.N, and Tb.Th for distal femurs.
Osteoclast differentiation assay
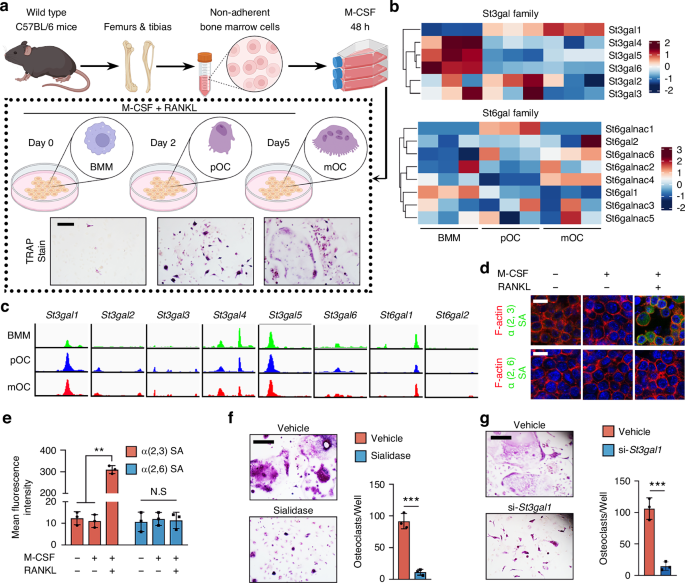
For TRAP staining, BMMs were seeded in 96-well plates (5 × 103 cells/well) and cultured with M-CSF (50 ng/mL) and RANKL (100 ng/mL). At 0, 24, and 96 h post-stimulation, cells were fixed with 4% paraformaldehyde for 20 min, then stained with TRAP solution (0.1 mg/mL naphthol phosphate, 0.3 mg/mL Fast Red Violet) following the manufacturer’s protocol. TRAP activity was analyzed by colorimetry. For immunofluorescence (IF), BMMs were cultured similarly for 4 days with 1 U/mL SialEXO 23 α2-3 sialidase and 10 μmol/L U0126, then fixed, permeabilized with 0.2% Triton X-100, and blocked. Cells were stained with vinculin antibody (1:500) for 1 h at 37 °C and counterstained with DAPI (1:1 000) for 10 min before fluorescence and confocal microscopy. For the pit formation assay, BMMs were seeded in 96- or 48-well Corning Osteo Assay Surface plates (2 × 103 or 1 × 104 cells/well) and induced with RANKL and M-CSF for 5 days. Cells were removed with bleach solution, and pit formation area was analyzed as previously described.
Immunohistochemistry, immunofluorescence, and histomorphometry
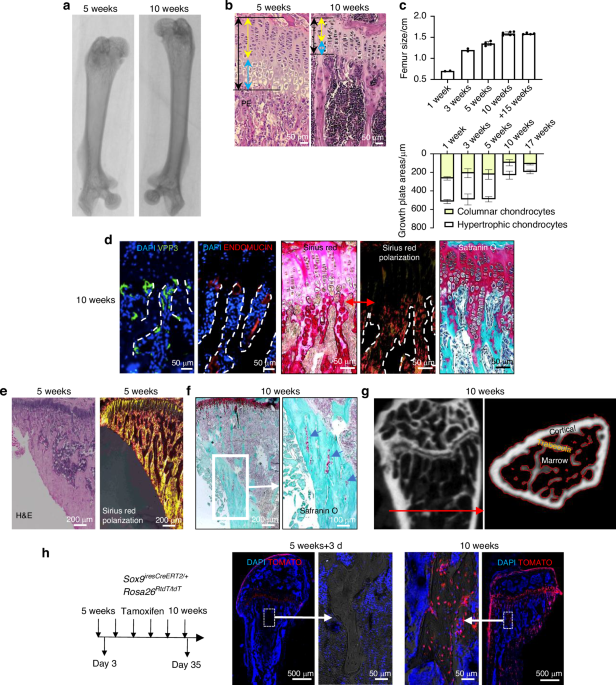
Femurs were collected, fixed in 4% paraformaldehyde overnight, and decalcified in 10% EDTA (pH 7.4) for 21 days. Samples were dehydrated in 30% sucrose for 24 h and embedded in paraffin or optimal cutting temperature compound (Sakura Finetek). Four-micrometer-thick coronal sections were prepared for hematoxylin and eosin staining. For immunostaining, femurs were fixed for 4 h in 4% paraformaldehyde, decalcified in 0.5 mol/L EDTA at 4 °C for 24 h, and dehydrated in 20% sucrose and 2% polyvinylpyrrolidone (PVP) for 24 h before embedding in 8% gelatin. Forty-micrometer-thick coronal sections were processed for immunostaining using primary antibodies against ST3GAL1 (1:50) and TRAP (1:100) at 4 °C overnight. Immunohistochemical detection was performed with a horseradish peroxidase-streptavidin kit, followed by hematoxylin counterstaining. Sialic acid detection was performed using biotinylated Maackia amurensis lectin II (MAL II) to detect α2,3-linked sialic acids and Sambucus nigra lectin (SNA) to detect α2,6-linked sialic acids, followed by fluorescein-conjugated streptavidin for signal visualization. Imaging was conducted using a Zeiss LSM 780 confocal or Olympus BX51 microscope, and quantitative histomorphometry was performed using OsteoMeasure XP software in a blinded manner.
ChIP-PCR assay
For the ChIP assay, BMMs were cultured with M-CSF and RANKL for 72 h to detect ST3Gal1 core enhancer DNA binding. Cells were lysed and cross-linked with 1% formaldehyde, followed by DNA fragmentation via micrococcal nuclease (MNase) digestion. Ten percent of the sample was preserved as input, and the remaining supernatant was incubated overnight at 4 °C with ChIP Grade c-FOS antibody (Abcam, ab27793, 10 μg). RNA Polymerase II antibody served as the positive control, and normal Rabbit IgG was the negative control. Immunoprecipitation was performed using ChIP Grade Protein A/G Magnetic Beads (Thermo Fisher Scientific, 26157). Eluted DNA was purified according to the manufacturer’s instructions and prepared for PCR detection.
Luciferase reporter assay
For the Luciferase reporter assay, wild-type and mutant (TGACTCA to ATATCGT) ST3Gal1 enhancer regions were cloned into pGL3-Basic, co-transfected with pRL-TK into RAW264.7 cells using Lipofectamine 3000, and treated with RANKL (50 ng/mL) for 48 h. Luciferase activity, normalized to Renilla, was then detected.
Bioinformatics
Gene core enhancer transcription factor binding was predicted using the ENCODE project and ChIP-seq data from the UCSC Genome Browser (https://genome.ucsc.edu/ENCODE). RNA-seq raw and processed data have been deposited in the GEO database under accession number GSE133515.
Immunoblotting analysis
Cells were lysed in IP buffer (50 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl, 1% Triton X-100, 0.5% sodium deoxycholate) with protease inhibitors. Lysates were immunoprecipitated using primary antibodies against TLR2 (Santa Cruz Biotechnology, sc-21759) and Siglec15 (Thermo Fisher, PA5-48221), followed by Protein A/G absorption (Thermo Fisher, 26149). Immunoprecipitates were separated by SDS-PAGE, transferred to nitrocellulose, and detected using the SuperSignal West Femto Substrate system (Thermo Fisher). For immunoblotting, cells were lysed in IP buffer with 0.5% SDS and incubated overnight at 4°C with primary antibodies against p-ERK, ST3GAL1, c-FOS, ER-alpha, TRAF6, and Siglec15 (1:1 000). Secondary antibody incubation was performed for 1 h (1:2 000).
Human subjects, serum sialic acid measurement, and scRNA-seq
The study cohort comprised pre-menopausal women (n = 10), post-menopausal women without osteoporosis (n = 10), and post-menopausal women with osteoporosis (n = 10), with serum samples collected from all participants. Total sialic acid levels were quantified using a commercial colorimetric assay, and α-2,3-linked SA levels were measured by a lectin-based ELISA employing Maackia amurensis lectin II (MAL-II), which specifically recognizes α-2,3-linked sialic acids. Bone and bone-marrow specimens were obtained from four post-menopausal osteoporotic (postMP-OP) patients (ages 65–73) undergoing spinal surgery and four pre-menopausal (preMP) women (ages 24–42, GSE120221), after informed consent and institutional ethics approval.
For scRNA-seq, freshly resected bone and marrow tissues were stored in tissue-preservation solution (Miltenyi Biotec) until processing. Samples were washed with PBS, cut into small pieces, and digested at 37 °C for 50 min with Collagenase II (150 U/mL), Collagenase IV (2 mg/mL), Dispase II (1.2 U/mL), and DNase I (50 U/mL). The digests were filtered through a 70 µm cell strainer and centrifuged at 300 × g for 5 min. After red-blood-cell lysis (Miltenyi Biotec), cells were washed in PBS containing 0.04% BSA, passed through a 35 µm filter, and loaded onto the 10x Genomics Chromium Controller for single-cell capture using the Single Cell 3′ v3 reagent kit. Libraries were sequenced on an Illumina 150 bp paired-end run, and reads were aligned to the human reference genome (GRCh38) with CellRanger v3.1.0 to generate the single-cell barcode matrix. Cell types were defined from their gene-expression profiles using established marker genes for bone and immune cells (see Supporting Information). All downstream analyses, including Seurat-based integration and donor-level batch correction, as well as differential-expression and GSEA analyses, were performed in R (RStudio).
Gene Ontology (GO) and KEGG pathway enrichment analysis
Differential expression analysis was carried out on donor-level pseudobulk profiles generated from the scRNA-seq data. For each clinical group (pre-menopausal, post-menopausal non-osteoporotic, and post-menopausal osteoporotic) and for each annotated cell type, we identified differentially expressed genes (DEGs) using a donor-adjusted one-versus-rest model implemented in the voom/limma pipeline. Genes with false discovery rate (FDR) < 0.05 and |log₂ fold change| > 0.25 were considered significant. Significant up- and down-regulated DEGs from each cell type were then used as input for Gene Ontology (GO) Biological Process and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses using the clusterProfiler R package (v4.0). Enrichment results were expressed as adjusted P values (Benjamini–Hochberg correction).
Statistical analysis
Data represent at least three independent experiments performed in triplicate unless stated otherwise. Statistical analysis was conducted using Prism 7.0 (GraphPad), with specific methods detailed in the figure legends. P values < 0.05 were considered significant, and error bars represent standard deviation (SD).

Comments (0)