Animals and PBMT treatment in vivo
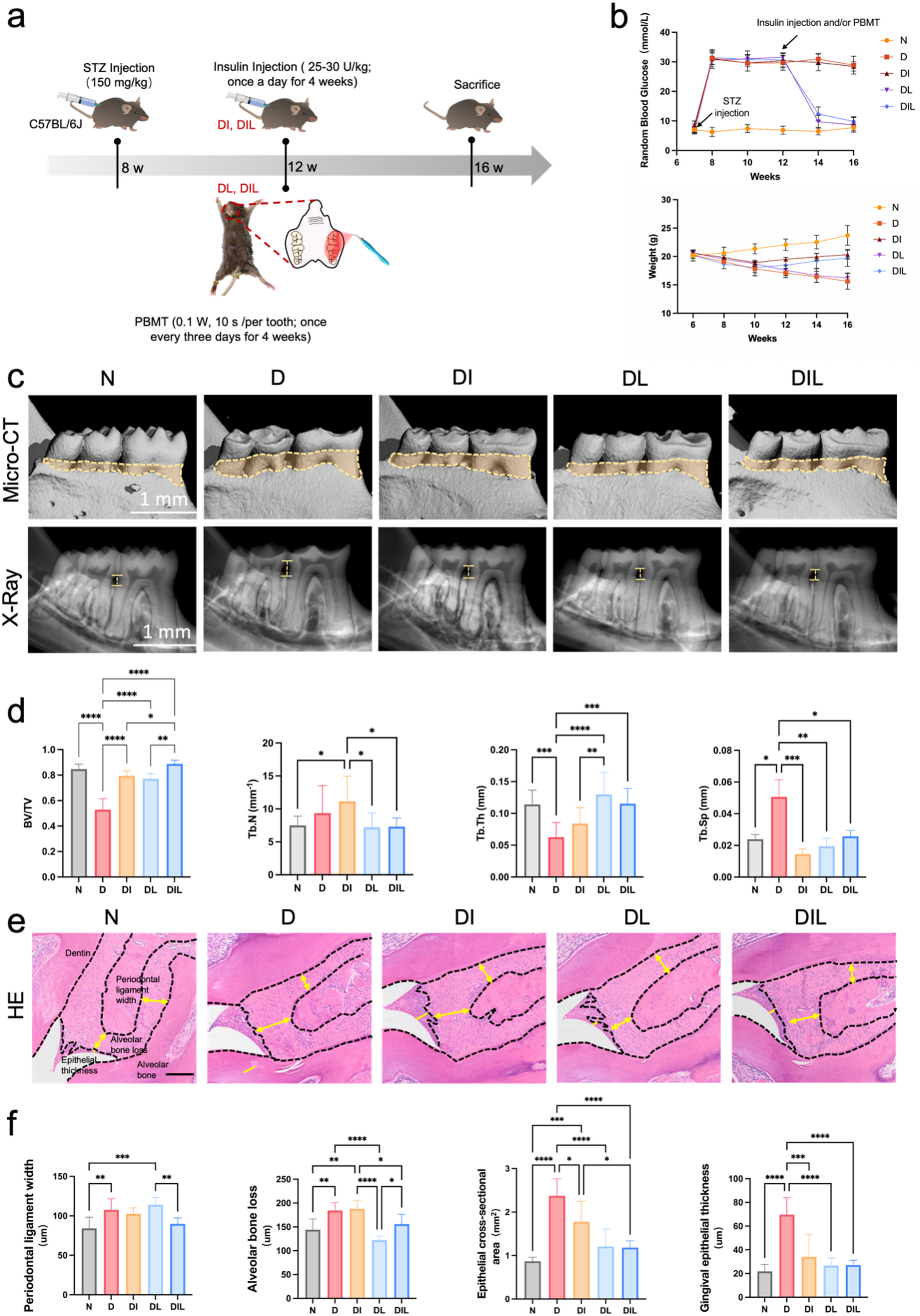
Six-week-old male C57BL/6J mice (Nanjing Model Animal Center, Nanjing, China) were housed at our centre under approved protocols (WCHSIRB-D-2024-586) and randomly allocated to five groups (n = 6 per group): control (N), diabetic (D), insulin-treated (DI), PBMT-treated (DL), and combined therapy (DIL). DM was induced through intraperitoneal injection of streptozotocin (STZ; 150 mg/kg in pH 4.5 buffer), with diabetic status being confirmed by the blood glucose levels exceeding 16.7 mmol/L [25]. Starting at week 12, the DI and DIL groups received daily insulin injections (25–30 U/kg), while the DL and DIL groups underwent PBMT using a 980 nm laser at 0.1 W power and 3 J/cm² energy density (10 s per tooth, twice weekly for 4 weeks). The irradiation spot area was calculated as 0.33 cm² based on the energy density formula, with a working distance of 1.4 cm. Control mice received equivalent volumes of saline [26, 27].
Cell culture and PBMT treatment in vitro
iBMDMs (Fu Heng Biology) and RAW 264.7 macrophages (State Key Laboratory of Oral Diseases, Chengdu, China) were maintained in high-glucose Dulbecco’s modified Eagle medium (DMEM, Gibco, USA) containing 10% foetal bovine serum (FBS, Gibco, USA) under standard culture conditions (37 °C, 5% CO₂).
iBMDMs were allocated into distinct groups. The control group (NG) received DMEM with 25 mM glucose. Hyperglycemic (HG) conditions were established using 45 mM glucose-containing DMEM for 6 days. The HG-NG transition group experienced initial exposure to 45 mM glucose for 3 days followed by 3 days in 25 mM glucose medium.
For PBMT treatment, cells cultured in 12-well plates (base area ≈ 4.4 cm²) were first maintained under high-glucose conditions for three days, then transferred to low-glucose medium and irradiated using a 980 nm semiconductor laser system (GigaaMedical, China). Based on the formula Energy Density = Power × Time/Spot Area, three energy densities (3, 6, and 18 J/cm²) were achieved using power outputs of 0.1, 0.2, and 0.6 W respectively, with a constant irradiation time of 132 s and working distance of 5 cm.
In the inhibitor experiments, cells were pretreated with specific inhibitors for 1 h prior to exposure to high-glucose (45 mM) conditions for 3 days, followed by 3 days in low-glucose (25 mM) medium. The inhibitors included: 1 µM AS1842856 (FOXO1 inhibitor, Ambeed, USA), 2 µM AKT inhibitor VIII (Ambeed, USA), and 10 µM FR 180,204 (ERK inhibitor, Ambeed, USA). For combination treatments, inhibitor-pretreated cells undergoing the same glucose transition received 980 nm laser irradiation at 0.1 W for 132 s during the low-glucose phase.
Western blot (WB)
Proteins were extracted from iBMDM cells or murine gingival tissues using RIPA lysis buffer supplemented with protease and phosphatase inhibitors. Cell lysates were centrifuged (14,000 × g, 10 min), and tissue samples were homogenized in liquid nitrogen, followed by sonication and centrifugation. Supernatants were mixed with 5× SDS loading buffer, denatured (100 °C, 6 min), and separated by SDS-PAGE. After transfer to PVDF membranes, blots were incubated overnight at 4 °C with primary antibodies (β-actin 1:10,000; β-tubulin 1:10,000; IL-1β 1:1,000; TNF-α 1:2,000; HK2 1:1,000; GLUT1 1:2,000; PKM2 1:1,000; DNMT1 1:4,000; FOXO1 1:5,000; AKT/p-AKT 1:2,000; ERK/p-ERK 1:5,000; MEK 1:5,000), followed by 1-h incubation with HRP-conjugated secondary antibodies (1:50,000). Signals were detected using enhanced chemiluminescence (ChemiDoc MP, Bio-Rad).
Micro-computed tomography (CT) assay
A µCT50 system (Olympus, Japan) was utilised to scan mandibular bones, followed by 3D reconstruction and bone density analysis using SCANO Visualiser (https://scano.app).
Haematoxylin and eosin (H&E) staining and immunostaining
Mouse maxillae were fixed in 4% paraformaldehyde (PFA, Servicebio, China), decalcified in ethylenediamine tetraacetic acid, and embedded in paraffin for sectioning. H&E staining was performed using standard protocols. For immunohistochemistry, sections underwent antigen retrieval, peroxidase blocking, and incubation with primary antibodies followed by HRP-conjugated secondary antibodies, with DAB used for chromogenic development. For immunofluorescence, samples were processed similarly and incubated with primary antibodies against HK2 (1:200), PKM2 (1:250), ERK (1:50), p-ERK (1:100), F4/80 (1:200), and DNMT1 (1:200), followed by fluorochrome-conjugated secondary antibodies (Cy3 1:500, FITC 1:100). All images were acquired using confocal microscopy (Olympus, Japan).
Transwell migration assay
Cells were resuspended in DMEM devoid of serum components and placed in the upper compartments of the migration chambers, whereas the lower compartments received medium enriched with 20% FBS. After a 12-h incubation, celluar fixation was performed using 4% PFA, followed by staining with 0.1% crystal violet for 30 min and PBS washes; subsequently, the non-migrated cells were removed from the upper membrane. The migrated cells were quantified using a bright-field microscope (Olympus, Japan).
Senescence-associated β-galactosidase (SA-β-GaL) staining
Cellular senescence was evaluated through SA-β-GaL staining kit (Beyotime, China). RAW 264.7 underwent fixation with 4% PFA for 15 min at ambient temperature. After PBS washing, cells were treated with staining solution and maintained at 37 °C under light-protected conditions for 16 h. Following additional PBS rinses, stained specimens were examined microscopically, with quantitative analysis of positively stained cells performed using ImageJ software (NIH, USA).
Latex bead phagocytosis assay
The red fluorescent latex beads (Sigma-Aldrich, USA) were prepared by diluting in 1% bovine serum albumin (1:100 ratio), followed by a 30-minute incubation at 37 °C and 5-minute sonication. Subsequently, 2 mL of the prepared bead solution was introduced into the culture medium and maintained at 37 °C for 90 min. Following incubation, cellular uptake was quantified through flow cytometric, with data processing performed using FlowJo software (FlowJo, USA).
Flow cytometry
iBMDMs were collected and adjusted to a concentration of 1 × 106 cells per sample. For intracellular FOXO1 detection, cells were stained with FOXO1 antibody (Proteintech, USA) followed by FITC-conjugated secondary antibody (Beyotime, China). For macrophage polarization assessment, cells were stained with PE-Cyanine7 anti-CD86 (eBioscience, USA) and PE anti-CD206 (eBioscience, USA). All samples were analyzed using a CytoFLEX S flow cytometer (Beckman, USA).
ATP and LDH content assay
ATP and LDH levels were measured using an ATP assay kit (Beyotime, China) and LDH assay kit (Solarbio, China). Standard curves were constructed, followed by iBMDM lysis on ice, sonication, and centrifugation. The supernatants were mixed with detection reagents, incubated at 37 °C for 30 min, and analysed using a microplate reader (ATP: 340 nm; LDH: 450 nm). The concentrations were calculated using standard curves.
Statistics
Each experimental procedure was replicated a minimum of three times independently. The analysis of data was conducted utilizing GraphPad Prism (GraphPad Software, USA), with results presented as mean values accompanied by standard deviations. For comparisons involving multiple groups, one-way ANOVA followed by Tukey’s post-hoc test was employed, while unpaired Student’s t-tests were utilized for two-group analyses. A threshold of P < 0.05 was established to determine statistical significance.


Comments (0)