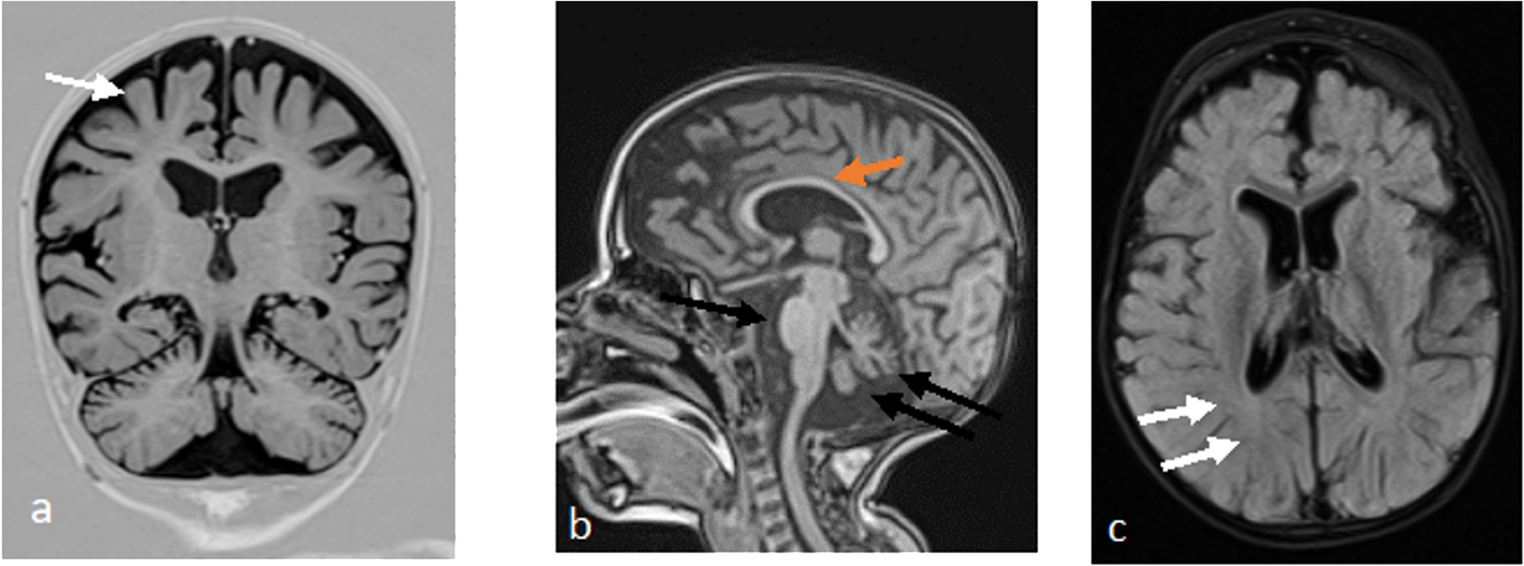
Suleiman–El-Hattab syndrome (SULEHS; OMIM #618674) is an ultra-rare autosomal recessive multisystem neurodevelopmental disorder caused by biallelic loss-of-function variants in TASP1, with a characteristic combination of global developmental delay/intellectual disability, distinctive craniofacial dysmorphism, recurrent infections, congenital anomalies, and a typically cheerful affect. Because fewer than ten molecularly confirmed individuals have been reported, the phenotypic spectrum and neuroradiologic variability remain incompletely defined. We describe the first genetically confirmed Iranian patient with a clinical phenotype suggestive of SULEHS, including microcephaly, failure to thrive, syndromic facial features, neurodevelopmental delay (NDD), recurrent pneumonias, cardiac and genitourinary anomalies, hearing impairment, seizures, and a happy demeanor with drooling. Brain MRI demonstrated ventriculomegaly and a posterior fossa malformation consistent with a Dandy–Walker variant. Conventional cytogenetics and array-CGH were uninformative; therefore, whole-exome sequencing (WES) with segregation analysis was performed. In parallel, we conducted a targeted literature review and systematically compared our patient’s clinical and imaging findings with eight previously reported molecularly confirmed cases. WES identified a novel homozygous TASP1 variant (NM_017714.3:c.358 A > G; p.Met120Val), classified as a variant of uncertain significance (VUS), with both parents confirmed as heterozygous carriers. Comparative analysis across the eight published cases and our patient demonstrated substantial overlap in core features—microcephaly, failure to thrive, typical craniofacial gestalt (e.g., thick highly arched eyebrows with synophrys, hypertelorism, periorbital fullness, ear anomalies), recurrent respiratory infections, cardiovascular anomalies, developmental delay/intellectual disability (DD/ID), happy demeanor, and drooling—while several commonly reported findings (e.g., epicanthus, thick eyelids, thick lower lip vermilion, lumbosacral hirsutism, feeding difficulties, hypotonia) were absent in our patient. Neuroimaging across prior cases included corpus callosum abnormalities, ventriculomegaly, encephalomalacia, and posterior fossa malformations; in our case, only ventriculomegaly and posterior fossa malformation were present. This report expands the geographic and clinical spectrum of SULEHS by documenting the ninth reported molecularly confirmed case and the first from Iran, and it further delineates phenotypic and neuroradiologic heterogeneity by showing ventriculomegaly with a posterior fossa malformation consistent with a Dandy–Walker variant. In our patient, conventional genetic testing (karyotype and array-CGH) was uninformative, whereas WES identified a novel homozygous TASP1 classified as VUS with parental carrier segregation; although functional validation is still required, the strong phenotype–genotype concordance supports its likely contribution and underscores the diagnostic value of comprehensive genomic testing in undiagnosed neurodevelopmental syndromes.


Comments (0)