
Remember me
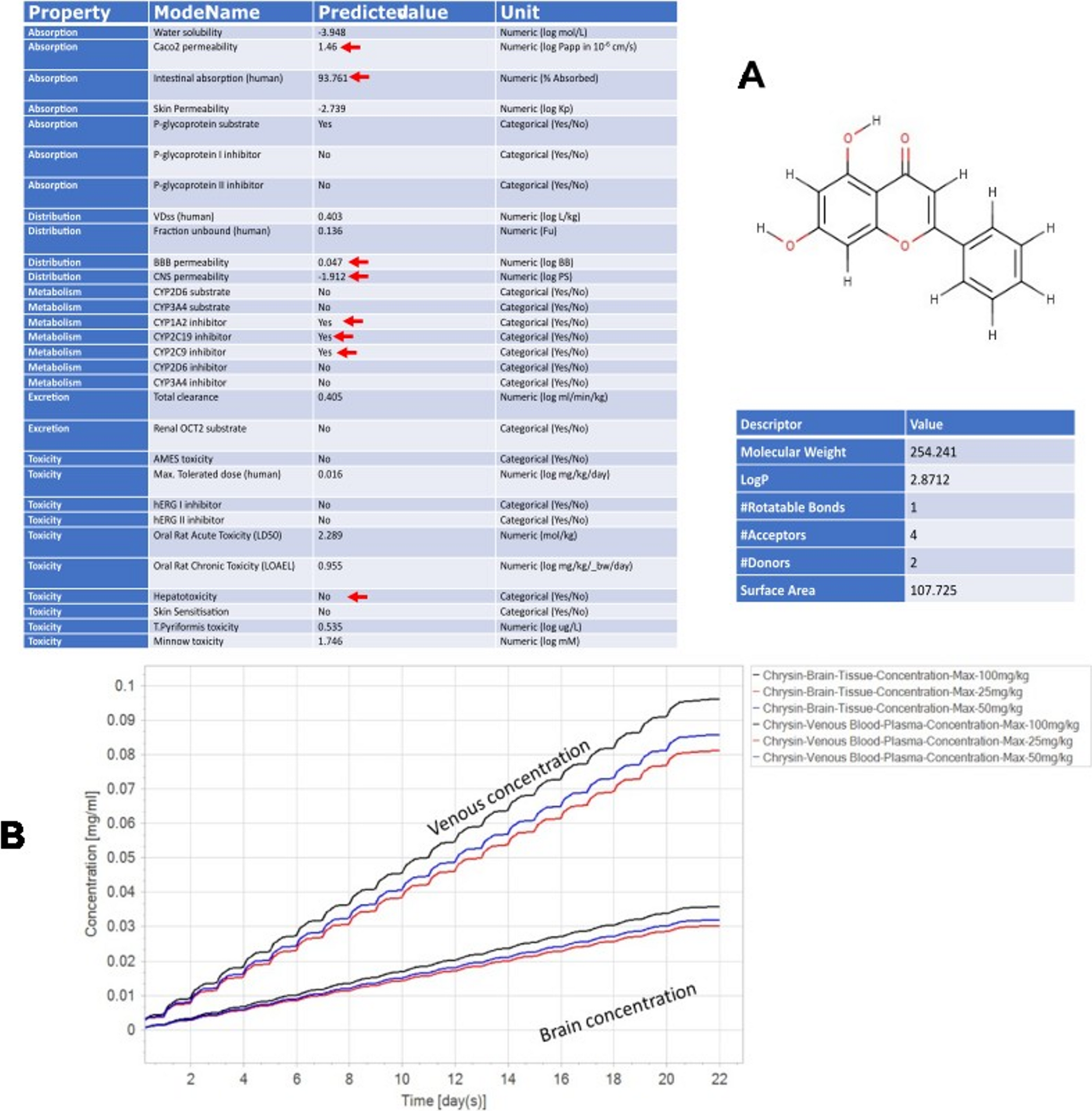

A total of 385 articles were initially identified. Of these, 60 studies were selected after excluding 238 duplicates or irrelevant articles. Following the full-text review, a total of 23 articles were selected for this study after excluding 37 studies for reasons outlined in the PRISMA diagram, such as outcomes not relevant to this study or ASD. The PRISMA flow diagram depicting this study is shown in Fig. 3.
Fig. 3
PRISMA flow diagram depicting study. This figure represents a PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) flow diagram, showing the process of study selection discussed in this systematic review
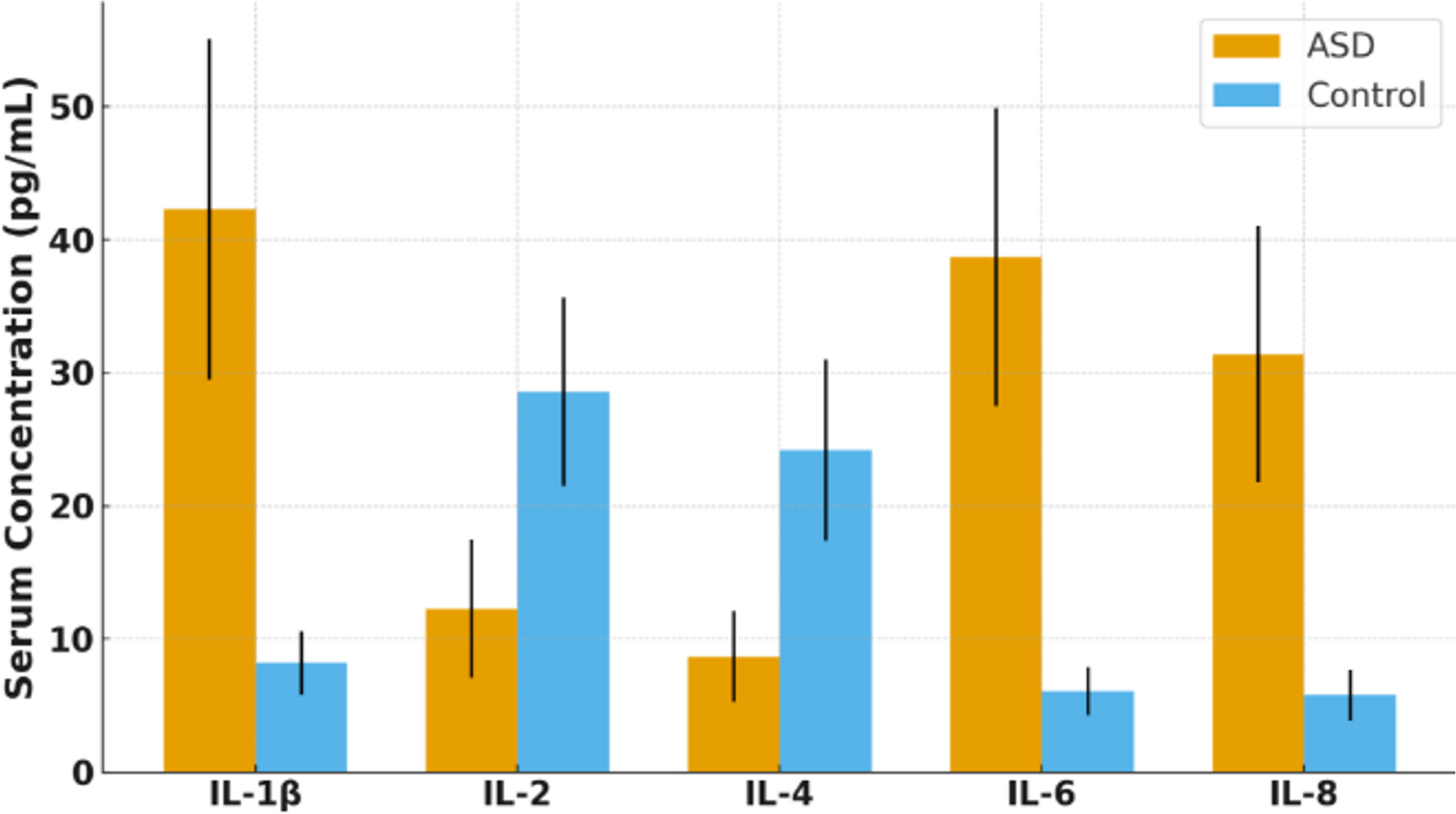
Quality Assessment of Included StudiesThe critical review of the studies included in this systematic review is shown in Fig. 4.
Fig. 4
Risk of bias assessment for included preclinical studies evaluating purinergic signaling in autism spectrum disorder. Each study was assessed across nine domains using the SYRCLE risk of bias tool. Green circles indicate low risk of bias, yellow circles indicate unclear risk, and red circles indicate high risk. D1–D9 correspond to sequence generation, baseline characteristics, allocation concealment, random housing, blinding of caregivers/investigators, random outcome assessment, blinding of outcome assessment, incomplete outcome data, and selective outcome reporting
Data Extraction and Study OutcomesA summary of the main findings of studies included in this review article is shown in Table 1. The studies included in this review reflect different aspects of autism in rodents and zebrafish and can be broadly categorized into:
1)Genetic modifications: involving mutations in specific genes known to be associated with ASD in humans (12 studies)
2)Environmental exposures:involving prenatal or postnatal exposure to environmental risk factors on neurodevelopment (11 studies)
Table 1 Summary of preclinical studies on purinergic signaling in autism spectrum disordersStudies on Genetic Modifications and Purinergic Signaling DysregulationsFragile X messenger ribonucleoprotein 1 (Fmr1)—Knockout (KO), (Fmr1-/-) mouse model (6 Studies): Mutations in the FMR1 gene lead to Fragile X syndrome (FXS), a monogenic disease with ASD phenotypes including neurodevelopmental and intellectual disabilities as well as neuro inflammatory symptoms (Richter and Zhao 2021). The Fmr1-/- mouse model is used to study FXS and ASD (Bernardet and Crusio 2006). The most important behavioral alterations observed in the Fmr1-/- mouse are social deficits, increased repetitive behavior, anxiety and hyperactivity (Bernardet and Crusio 2006).
In 2021 Reynolds et al. sought to understand if astrocyte-mediated purinergic signaling is dysregulated in Fragile X Syndrome (FXS) (Reynolds et al. 2021b). This is an important issue, since purinergic signaling is one of the most ubiquitous signaling systems for glial-neuronal and glial-glial crosstalk. The study showed that in the presence of exogenous ATP and UTP the intracellular calcium responses were elevated in Fmr1 KO astrocytes compared to wild-type (WT) controls. Furthermore, the expression of purinergic P2Y2 and P2Y6Rs was elevated in Fmr1-/- astrocytes, both in vitro and in dissociated cortical tissues. Together these findings point to an upregulation of purinergic signaling pathways in the absence of the Fmr1 gene. Suramin, a nonspecific P2Y antagonist, normalized this response. The expression and secretion of Thrombospondin-1 (TSP-1) a synaptogenic protein regulated by P2YR activation, also increased in Fmr1-/- astrocytes while its levels were transiently elevated in the cortex of the Fmr1-/- mouse. In summary, activation of P2Y2/P2Y6R-mediated purinergic signaling in Fmr1-/- mouse cortical astrocytes promoted aberrant excitatory synaptic transmission in the cortex. Targeting this cortical purinergic signaling may offer a potential therapeutic approach to restore normal excitability and improve cognitive and behavioral outcomes in affected individuals with FXS and ASD.
In a follow up study in 2021 Reynolds et al. investigated the mechanisms underlying elevated interleukin (IL)−6 secretion in cortical astrocytes from the Fmr1-/- mouse (Reynolds et al. 2021a). The group explored how purinergic and immune pathways coordinated to secrete IL-6, a pro-inflammatory cytokine important for neuroinflammation and synaptic alteration. The study demonstrated that activation of glial P2Y purinergic receptors enhanced the secretion of glycoprotein tenascin C (TNC) and promoted phosphorylation of transcription factor, signal transducer and activator of transcription 3 (Stat3). Phosphorylated Stat3 (pStat3) increased gene expression of IL-6 and exacerbated pro-inflammatory responses in the FXS-mouse cortex. Stat3 knockdown and Toll-like receptor 4 (TLR4) antagonism (with TAK242) normalized IL-6 release, suggesting that purinergic and immunological pathways converge to drive IL-6 secretion via Stat3. A relatively recent study by the same group showed that upregulated purinergic signaling in cortical FXS astrocytes increases neural firing rates (Reynolds et al. 2024a). The increased firing was, however, associated with reduced synchrony, suggesting a disruption in coordinated neural activity. These findings underscore the significance of astrocyte-mediated purinergic signaling in the development of neural circuitry and point to its potential role in the pathophysiology of FXS. Given the frequency of synaptic dysfunction observed in ASD, such preclinical studies focusing on the effects of purinergic signaling on synaptic functioning and neurodevelopment are of paramount importance. A selective P2Y2R antagonist, AR–C 118925XX further restored this aberrant neural activity in Fmr1-/- neural cultures, highlighting that P2Y2 is a potential therapeutic target for FXS. Targeting P2Y2R-mediated purinergic signaling pathways may, therefore, offer a promising therapeutic strategy for developmental intellectual disabilities associated with ASD.
In a separate study in 2024, Napier et al. investigated the role of purinergic P2X7R’s in hippocampal brain development in the FXS mouse model (Napier et al. 2024). Their research showed that P2X7 expression was decreased in the hippocampus of Fmr1-/- mouse at postnatal (P) days 14 and 21, critical for neurite outgrowth and synaptic refinement. This suggests that altered hippocampal P2X7 expressions during postnatal stages may affect brain development in the FXS mouse. Immunohistochemical analysis further revealed increased expression of P2X7Rs by Fmr1 KO microglia, indicating glial inflammation and a shift in purinergic signaling within the hippocampal microenvironment. This study also found sex-dependent variations in P2X7R expression. Male Fmr1-/- mice exhibited reduced colocalization of P2X7R with the neuronal cells at P14 and P21 compared to females. Conversely, only female Fmr1-/- mouse showed a reduction in neuronal P2X7R expression in the dorsal hippocampus, highlighting the importance of considering sex as a variable in purinergic signaling and autism research.
In the same year, Reynolds et al. reported dysregulation in the extracellular concentration of adenosine-based purinergic molecules like UDP, ATP, AMP, and intracellular adenosines in Fmr1-/- astrocytes (Reynolds et al. 2024b). Conditioned media from KO astrocytes had a higher concentration of secreted adenosine. The Fmr1-/- astrocytes also had higher levels of active glycosylated membrane-bound CD39 ectonucleotidases. Alterations in their level, together with a change in the adenosine-based purine metabolites in Fmr1-/- astrocytes, points to an impairment in purine metabolism and signaling which could contribute to the synaptic and network dysfunctions characteristic of FXS.
Taken together, the above two studies demonstrate that absence of astrocyte Fragile X messenger ribonucleoprotein (Fmrp) in a preclinical ASD-model (Fmr1-/- mouse), leads to widespread purinergic dysregulation affecting not only receptor expression but also the production and availability of several purines which are associated with improper brain development in ASD.
A study published by Ferrante et al. in 2021 described the interaction of adenosine A2A receptors (A2ARs) and Fmrp in the cortex and hippocampus of Fmr1 WT/KO mice suggesting that Fmrp regulates A2AR expression during early neurodevelopment (Ferrante et al. 2021). Extracellular electrophysiology experiments in brain slices of Fmr1-/- mouse showed that A2ARs and metabotropic glutamate 5 receptors (mGlu5Rs) exhibit a synergistic interaction in the hippocampus, which is enhanced in Fmr1-/- mice. A2AR agonists and antagonists modulated hippocampal mGlu5R-induced synaptic effects/depression in Fmr1 WT and KO.
mice and the ability of the A2AR agonist CGS21680 to modulate this interaction was greater in absence of Fmrp, suggesting a greater synergistic coupling between A2AR and mGlu5Rs in the Fmr1-/-. Furthermore, the pharmacological blockade of A2ARs by the orally available antagonist, istradefylline, reduced the synaptic alterations and dendritic spine density in Fmr1-/- mouse. Learning and memory deficits in Fmr1-/- were also corrected by istradefylline, probably through synaptic modifications and reduction in dendritic spine density. This beneficial effect of istradefylline is encouraging, since FXS patients suffer from cognitive disabilities. Apart from these behavioral effects, the study also delineated the downstream mechanistic pathway of the A2ARs. Istradefylline ameliorated mTOR/mGlu5R hyper-phosphorylation and restored levels of the truncated form of tropomyosin receptor kinase B (TrkB). Truncated TrkB are important regulators of impaired BDNF signaling and have been identified as pivotal contributors to the development of several neurological disorders. These receptors lack intrinsic tyrosine kinase activity, resulting in signaling mechanisms that differ from those of their full-length counterparts (Tessarollo and Yanpallewar, 2022). The full-length TrkB protein supports signaling for brain-derived neurotrophic factor (BDNF) and striatal-enriched protein tyrosine phosphatase (STEP) within the hippocampus. Collectively, these findings suggest that istradefylline exerts its effects via mTOR/mGlu5R and TrkB/BDNF pathways. RNA immunoprecipitation studies further showed that A2AR-mRNA is associated with the RNA-binding protein, Fmrp, in a complex in the cortex and hippocampus. This suggests that during early stages of neurodevelopment, Fmrp could regulate translation of A2AR mRNA. Therefore, abnormalities related to A2AR deficiency could start early in development in Fmr1-/- mice. Overall, this study demonstrated that blocking the purinergic A2AR, could restore mGlu5R-mediated effects in FXS including synaptic and cognitive alterations associated with this condition.
LimitationsThe Fmr1-/- mouse model typically uses a full genetic knockout while in humans frequently there is a silencing of the gene that leads to a deficiency of the protein, rather than a complete absence (Bakker and Oostra 2003). So, the relevance of findings in this model to human FXS must be interpreted with caution. The reported studies lack vivo/across model validation and focus on one cell type/pathway, with challenges in quantifying ligand levels, metabolites and limited functional, behavioral linkage. They capture a snapshot rather than full developmental dynamics. These constraints limit generalizability to the whole intact brain and full developmental trajectory of purinergic signaling in this model.
BTBR T + Itpr3tf/J (BTBR) mouse model (2 Studies): The BTBR mouse, originally bred for diabetic research studies, has deletions in the inositol triphosphate receptor 3 gene (Itpr3) which affects its social communication to food-preference. This mouse displays strong and consistent autism-related behaviors broadly classified under (1) Social deficits like impaired social interactions, less social play and poor performance in three-chamber social interaction tests. (2) Communication deficits demonstrated by reduced ultrasonic vocalizations and altered vocal pattens during social interactions (3) Repetitive behaviors like increased self-grooming, repetitive jumping, anxiety and displaying behavioral inflexibility in tasks requiring cognitive flexibility. (4) Neurological and immune features displayed by altered brain morphology, errors in axon guidance and neurogenesis as well as synaptic and neuroimmune dysfunctions (Meyza et al. 2013). Considering its complex genetic, molecular and physiological background, the BTBR mouse represents an idiopathic form of autism and supports a model to study multiple aberrations found throughout the ASD population in one animal (Meyza and Blanchard 2017; Meyza et al. 2013).
Ansari et al. used the BTBR ASD mouse model to investigate if P1 purinergic A2AR regulation of neuroimmune function through the retinoid-related orphan receptor gamma (RORγt)/Th17 signaling pathway (Ansari et al. 2017b). The A2AR agonist (CGS 21680) and an antagonist (SCH 5826) were administered for 7 days to evaluate behavioral outcomes, such as self-grooming, repetitive behaviors, pain sensitivity, and immune responses. The study revealed that activation of A2ARs with CGS 21680 reduced pro-inflammatory markers IL-17A, RORγt, Stat3, and pStat3 and elevated anti-inflammatory markers [forkhead box P3 and IL-10] in both spleen CD4 + T cells and brain tissues of the BTBR mouse as compared to the C57BL/6 (B6) WT. Conversely, blocking A2ARs with SCH 5826 produced opposite effects. These results suggest that A2AR activation may have therapeutic potential in addressing the neuroimmune dysfunction frequently associated with ASD. This study also illustrates how purinergic dysregulation is associated with neuroimmune functions and behavioral alterations in ASD.
In another study Ansari et al. investigated if the A2AR signaling pathway regulates cytokine production in BTBR mouse (Ansari et al. 2017a), which was previously shown to exhibit elevated proinflammatory cytokines that modulate their behavioral response (Careaga et al. 2015). They examined if A2AR agonists and antagonists affect the levels of IL-2, IL-6, IL-9, interferon gamma (IFN-γ), tumor necrosis factor alpha (TNF-α), and transforming growth factor β (TGF-β) in spleen and in splenic CD4 T lymphocyte cells (CD4 T cells). mRNA and protein levels of these cytokines were also analyzed in the brain tissues. The results showed that SCH 58261, an A2AR antagonist, upregulated levels of pro-inflammatory cytokines, while CGS 21680, an A2AR agonist, restored the balance between pro- and anti-inflammatory cytokines in the BTBR mouse, indicating the potential therapeutic promise of CGS 21680 for the treatment of autistic disorders.
LimitationsAlthough these studies show that A2AR regulates the expression of pro and anti-inflammatory cytokines, they do not necessarily prove that these cytokine changes drive immunologic dysfunction or behavioral phenotypes. Therefore, translating these findings into human ASD will require more work.
C58 Mouse Model (1 Study)The C58 inbred mouse strain is an attractive model for repetitive behavior characteristic of neurodevelopmental disorders including ASD. Compared to C57BL/6 mouse, C58 mouse shows high rates of spontaneous hindlimb jumping and backward somersaulting. This makes the model useful for studies deciphering the etiology and pathophysiology of aberrant repetitive behaviors associated with ASD (Whitehouse et al. 2017).
A study by Lewis et al. explored the potential of P1-adenosine receptor agonists to normalize repetitive behaviors associated with neurodevelopmental disorders (Lewis et al. 2019). A combination of adenosine A1 (CPA) and A2AR (CGS21680) agonists were administered to C58 mice each day, for 7 days. These agonists are known to alter the firing frequency of dorsal striatal neurons in the indirect pathway of the basal ganglia. Neither agonist alone was effective; however, their combination significantly reduced repetitive behaviors in both male and female mice over a six-hour period in the 7-day treatment course, indicating a long-lasting effect. Agonist treatment increased the expression of c-Fos, a marker for neuronal activity, in the dorsal striatum, suggesting enhanced activation of the direct and indirect pathways of the basal ganglia, which might have restored the balance in neural activity. The above findings point to P1-purinergic adenosine receptors of the dorsal striatum as prospective therapeutic target for ASD-related repetitive behaviors.
LimitationsThe high levels of jumping/backward somersaulting behaviors in this model do not fully recapitulate the complexity of ASD-related human repetitive behaviors. Though the agonists here showed behavioral improvement, the study does not support a mechanistic link that drug combination restored indirect-pathway activation. Also, the longer‐term effects of treatment, its potential side‐effects and detailed sex analysis need to be done.
P2X4R-deficient Mouse Model (1 Study)P2X4R is a member of the P2X superfamily of ion channels. They are classified as ligand-gated ion channels since their opening is triggered by the binding of a ligand, and are typically expressed by neurons of the brain, microglia, some epithelial and endothelial cells and intracellular compartments like lysosomes. P2X4Rs are the most sensitive of all the P2XRs. They respond to nanomolar concentrations of ATP and play a role in pain signaling, synaptic plasticity and trigger inflammation in response to extracellular (e) ATP (Suurvali et al. 2017). The P2X4-deficient mouse exhibits a plethora of socio-communicative and neurodevelopmental abnormalities akin to other murine models of ASD (Wyatt et al. 2013).
Wyatt et al. investigated the behavioral and neurochemical consequences of P2X4R deficiency in mice (Wyatt et al. 2013). The authors recorded the behavioral responses in 3- to 5-month-old male P2X4R heterozygous (HZ), knockout (KO) and WT mice. Both heterozygous HZ and KO mice displayed reduced social interactions and decreased ultrasonic vocalizations in response to maternal separation, indicating deficits in social communication and sensorimotor deficits Additionally, they showed enhanced tactile sensitivity and significant changes in the expression of glutamate receptor subunits in brain regions such as the prefrontal cortex and hippocampus, including decreased NMDA receptor subunits (GluN2A and GluN2B) and increased AMPA receptor subunits (GluA1 and GluA2). No significant differences were observed in anxiety-related behaviors or locomotor activity between KO, HZ, and WT mice. Collectively these results suggest that P2X4R signaling plays a crucial role in a plethora of socio-communicative and sensorimotor functions that are often affected in ASD, making these receptors an attractive therapeutic target.
LimitationsBecause the model is a constitutive P2X4 KO, it is difficult to disentangle, if the observed behavioral deficits reflect lifelong adaptations rather than the acute impact of P2X4 on behavior in adulthood or solely on its deletion. Also, only one receptor subtype in one sex, under one set of housing/handling/assay conditions were studied here limiting translation potential. The authors appropriately caution here that mouse social/vocalization assays do not accurately recapitulate human socio-communicative impairments.
Neuroligin-4 (Nlgn4) KO (Nlgn4-/-) Mouse Model (1 Study)NLGN4, a postsynaptic cell-adhesion molecule is implicated in human ASD. The encoding gene, NL4 is mutated in rare cases of familial autism. Its mutation leads to discrete developmental brain disorders, especially in synaptic organization and remodeling (Jamain et al. 2003). Nlgn4 KO (Nlgn4-/-) mouse shows increased repetitive behavior and altered social interaction. These conditions are more pronounced in the males, mimicking the higher incidences of ASD in men (El-Kordi et al. 2013).
A study published in 2023, investigated how the loss of Nlgn4 affects microglial cells in the brain, with a focus on sex-specific differences (Guneykaya et al. 2023). Fourteen-week-old Nlgn4-/- male mice were treated with 17β-estradiol via drinking water every two days, for 6 weeks. Microglial structure, function and proteomic profile in male and female sexes in WT and Nlgn4-/- mice were examined, in the CA3 region of the hippocampus. Male Nlgn4-/- mice exhibited reduced microglial density, less complex morphology, and diminished responses to injury in conjunction with alternations in purinergic signaling in the hippocampal CA3 region. The study revealed a sex-specific impairment in P2Y12-purinergic signaling in Nlgn4-/- males. Proteomic analysis revealed altered energy metabolism in male microglia, highlighting sexual dimorphism in microglial phenotypes associated with ASD. Furthermore, there was an impairment in male specific gamma oscillations. Gamma oscillations are important for cognition and for modulating the morphology and function of microglia, which are important for synaptic pruning, phagocytosis of pathogens and regulation of inflammation in the brain. Finally, estradiol treatment to male Nlgn4-/- mice restored the altered microglial phenotypes and functions, suggesting potential for sex-specific therapeutic strategies.
LimitationsMicroglia do not express Nlgn. So, the changes reported here do not reflect direct microglial effects and are plausibly secondary to neuronal networks. How the cellular/physiological changes drive behavioral ASD features need further investigation. Importantly this study does not encompass the developmental windows critical for autism and focuses only on the CA3 region of the brain hippocampus. Though the sex-difference findings here are interesting, the underlying cause of female resilience needs further investigation.
Inositol 1,4,5-triphosphate Receptor Type 2 (IP3R2)-deficient Mouse Model (1 Study)Astrocytic activation occurs through increased cytoplasmic calcium signals, primarily mediated by IP3R2 (Cao et al. 2013). The IP3R2 gene is affected by rare de novo copy number variants in ASD patients (Gilman et al. 2011).
Wang et al. demonstrated the crucial role of astrocyte-derived ATP in modulating ASD-like behaviors in mice (Wang et al. 2021), using the IP3R2 null mouse (IP3R2-/-), (ii) astrocyte-specific IP3R2 conditional knockouts (cKOs) which were generated by crossing the Aldh1l1-CreER mice with the IP3R2loxp/loxp mice and (iii) eliminated IP3R2 expression in medial prefrontal cortex (mPFC) astrocytes by injecting adeno-associated virus with IP3R2-specific shRNA. Compared to WT, the IP3R2-/- mouse showed deficits in social interaction, increased repetitive behaviors and markedly decreased astrocyte somatic Ca2+ signals which remained unaffected by GPCR-agonist cocktails. These animals also exhibited enhanced exploratory behaviors and showed no change in anxiety levels or locomotor activity. Similar to the IP3R2-/- mice, cKO mice had behavioral abnormalities and Ca2+ signaling impairment, but no change in exploratory behaviors or anxiety or locomotion, indicating that IP3R2 deletion is responsible for two core ASD-like behaviors: reduced social interactions and increased repetitive actions. The astrocyte-specific selective deletion by shRNA produced no changes in cognitive impairment or anxiety or locomotion but showed significant alterations in social behavior compared to the IP3R2-/- mice. In the astrocytes from IP3R2-/- and IP3R2 cKO mice compared to WT, there was also a drastic reduction in the neurotransmitter ATP, hinting to the role of astrocyte-specific IP3R2. This change was only observed at the level of extracellular (e) ATP, while total ATP and intracellular levels remained unchanged. Reduced eATP release and elevated ecto-ATPase may have contributed to this. The P2Y1R antagonist MRS2500 inhibited ATP release in WT mice, showing P2Y1Rs involvement. Additionally, astrocytes treated with ARL67156 had higher ATP levels in IP3R2-/- mice than WT mice, indicating increased ecto-ATPase activity. Injecting ATP before behavior analysis resolved social interaction issues in IP3R2-/- animals but not repetitive behaviors. ATP corrected social deficits in IP3R2 cKO mice without changing social preference. ATPγS infusions in mPFC, improved social impairments in both IP3R2-/- and IP3R2 cKO mice, suggesting mPFC as a potential locus. These effects depended on neuronal P2X2Rs. Knocking down P2X2R in the mPFC, removed the positive effect of ATP and independently induced social deficits in WT mice. P2X2R knockdown also impaired transmission through the GABAA receptors (GABAAR), indicating that astrocyte Ca2⁺-induced ATP release enhances cortical GABA inhibition via P2X2Rs. Additionally, the GABAAR agonist clonazepam also rescued social deficits, suggesting impaired GABAergic transmission in the autism-like behaviors. In summary, an astrocyte-derived ATP deficiency led to autism-like behavior in IP3R2-deficient mice. Both synaptic and behavioral abnormalities in these mice were partially or completely normalized by ATP application and enhancing GABAergic transmission.
LimitationsATPγS infusion in mPFC did not rescue repetitive behavior, indicating that astrocyte derived ATP via IP₃R2 does not fully explain all ASD-like behaviors in this model. The rescue experiments are acute/short-term manipulations and don’t reflect if chronic modulation over development would change ASD-like phenotype. Furthermore, these investigations were performed in adult astrocytes rather than early/developmental stages crucial for ASD and does not focus on other regions of the brain beyond the mPFC or address sex-specific variations.
Studies on Environmental Exposures and Purinergic Signaling DysregulationsValproic Acid (VPA) (5 Studies)Prenatal exposure to VPA, an environmental risk factor and commonly used drug to treat epilepsy, mood disorders increase the risk of ASD in offsprings (Wood et al. 2015). This association is particularly pronounced when exposure occurs during critical periods of neurodevelopment during pregnancy (Ornoy et al. 2023). Therefore, prenatal exposure to VPA in animals, including rodents, zebrafish have become well-established and widely used preclinical models for studying ASD. These models often exhibit characteristics of idiopathic autism, including deficits in social interaction and communication, increased repetitive behaviors and anxiety with an epigenetic or environmental background (Cohen et al. 2019).
A study published in 2017 investigated how VPA could affect purinergic signaling in adult zebrafish (Zimmermann et al. 2017). The authors reported no change in ATP and ADP hydrolysis or cytosolic adenosine deaminase (ADA) activity. AMP hydrolysis activity increased by 12.5% and ecto-ADA activity decreased by 19.2%. The gene expression levels of the eATP hydrolyzing enzyme, ectonucleoside triphosphate diphosphohydrolase 8 (entpd8), ADA 2.1, and A2a.1 mRNA were increased. These results suggest that embryological exposure to VPA leads to long-term alterations in extracellular nucleotide metabolism, potentially contributing to social behavior deficits in ASD-models.
Babiec et al. also reported that prenatal exposure to VPA affects the expression and activity of purinergic receptors in the developing rat brain (Babiec et al. 2022). Pregnant rats received a single intraperitoneal (i.p.) injection of VPA (450 mg/kg body weight) on embryonic day (E) 12.5, coinciding with neural tube closure. The researchers then analyzed changes in the expression and activity of specific purinergic receptor subtypes in offsprings at embryonic (E) day 19, a critical window in prenatal brain development. Protein levels of adenosine receptors (A1, A2b, A3) and purinergic receptors (P2X2, P2X3, P2X7, P2Y1) changed significantly. These receptors influence progenitor cell proliferation, neuroanatomical changes, synaptic function, neuron migration and differentiation, dendrite and axon formation, as well as glutamate/GABA balance and overall neural excitability (Burnstock 2018). Furthermore, treatment with adenosine, ATP, and the metabolically stable ATP analog ATPƳS significantly increased intracellular Ca2+ ([Ca2+]i) in cortical neurons from VPA embryos. Stimulation with P2X2/3 receptor agonist α,β-methylene-ATP further elevated [Ca2+]i, indicating receptor hyperactivity, while BzATP (P2X7/4R agonist) and ADPβS (P2Y1/P2Y12R agonist) decreased [Ca2+]i, pointing to reduced activity. Critically, this study suggests that environmental teratogens like VPA can potentially disrupt purinergic signaling pathways in the developing brain during embryogenesis and thereby contribute to ASD pathogenesis.
A study published in 2020 by Hirsch et al. investigated the effects of a single dose of suramin, a non-selective purinergic antagonist, on behavioral and molecular alterations in VPA-induced rat model of ASD (Hirsch et al. 2020). Pregnant females received a single injection (i.p.) of 600 mg/kg VPA at day E12.5. Male offspring received a single i.p. injection of suramin, 20 mg/kg. Suramin administration restored sociability and reduced anxiety-like behavior. However, it did not improve deficits in reciprocal social interactions or normalize the heightened pain threshold observed in VPA-exposed rats. While suramin did not alter the VPA-induced upregulation of purinergic receptors P2X4 and P2Y2 in the hippocampus or P2X4 in the medial prefrontal cortex, it normalized elevated levels of IL-6, a pro-inflammatory cytokine associated with neuroinflammation in ASD. These results underscore the promise of suramin to address certain ASD-associated behavioral abnormalities.
In 2023 Babiec et al. published a study investigating how prenatal exposure of VPA alters purinergic receptor expression in the brain and affects glial cell morphology in a rat model of ASD (Babiec et al. 2023). Pregnant rats received a single intraperitoneal injection of VPA at embryonic day 12.5 and male offspring were analyzed at postnatal day (P) 52 (adolescence). The hippocampus and cortex were assessed for expression and localization of purinergic receptor mRNA and proteins respectively. The hippocampus of the preclinical ASD rats showed an increase in P2X1R levels, with a decrease in P2X7 and P2Y1Rs, while the cortex had a reduced P2X1R levels. Cortical microglial cell numbers and morphology were significantly altered, and IL-6 levels were elevated. The administration of a non-selective P2R antagonist partially mitigated the glial cell changes. These findings demonstrate that prenatal VPA exposure leads to region-specific alterations in purinergic receptor expression in adolescent brains, contributing to cortical microglial activation/neuroinflammation and underlines the importance of purinergic receptors as potential therapeutic targets for ASD. The administration of a non-selective P2R antagonist partially mitigated the glial cell changes.
In a study in 2024, Babiec et al. reported that the activation of P2 purinergic receptors led to synaptic alterations, behavioral changes and development of autism-like symptoms in the VPA-injected rat model (Babiec et al. 2024). Wistar female rats were i.p. injected with VPA (450 mg/kg body weight) at E12.5. At P52 the male VPA offspring received a single i.p. injection of the non-selective P2-purinoceptor antagonists PPADS and isoPPADS (12.5 mg/kg body weight). After 24 h. behavioral tests were recorded. Rats were sacrificed the next day after behavioral testings. Reverse phase high performance liquid chromatography (HPLC) analyzed changes in purine metabolism (ATP, ADP, AMP and adenosine) in the cerebrospinal fluid while quantitative real-time polymerase chain reaction (qRT-PCR) and western blots detected mRNA and protein levels in the brain’s hippocampus, cortex and cerebellum. VPA induced hyperactivation of purinergic signaling led to activation of mTOR kinase, the catalytic subunit of the mTORC1 complex responsible for synaptic proteins homeostasis and phosphorylation of downstream eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1). This led to dissociation of eukaryotic translation initiation factor 4E (eIF-4E), and translation of synaptic proteins such as synaptophysin, vesicle-associated membrane protein ½ (Vamp1/2) and development of autistic behaviors. Inhibition of purinergic P2Rs prevented synaptic and behavioral alterations. This study highlights the role of VPA as an environmental risk factor to alter purinergic signaling in association with induction of ASD.
LimitationsVPA-prenatal exposure is only one of many mechanisms associated with ASD and the clinical situation in humans is far more complex, so translational potential remains speculative. Additionally, one dose and/or one time-point of VPA exposure were used in the studies limiting understanding of dose–response, timing effects, and whether different gestational windows would yield different results. Also, causal link between purinergic signaling and behavioral deficits and ASD need to be explored. Suramin’s lack of impact on social interaction deficits and purinergic receptor expression suggests that additional studies are necessary to fully understand its therapeutic potential.
Maternal Immune Activation (MIA) using Agents like Poly(I:C) and Hyperpurinergia Mouse Model (5 Studies)The MIA model replicates a potential environmental risk factor for ASD during pregnancy (Ellul et al. 2023). In 1960, after the U.S. rubella epidemic, it was first proposed that there might be a direct link between neurodevelopmental disorders like ASD and infection during pregnancy (Chess 1971). A fair amount of research over the years indicates that MIA during first and second trimesters of pregnancy, rather than infection by itself enhances the risk of ASD in offsprings. The severity of the infection depends on the dose of injection and the timing of exposure during pregnancy (Hornig et al. 2018). Rodent models generated by administration of either double-stranded RNA [polyinosinic:polycytidylic acid poly (I:C), for mimicking viral infection], or lipopolysaccharide (LPS, for mimicking bacterial infection) have helped to elucidate the link between systemic maternal inflammation and MIA-induced neuroinflammation in ASD offsprings. The offsprings typically exhibit behavioral problems, such as reduced social interaction, anxiety and repetitive behaviors seen in ASD (Bao et al. 2022). Hyperpurinergia likewise plays a significant role in the development of ASD-like features. The hyperpurinergia model here investigates the effects of eATP in typical mice and the cumulative impact of hyperpurinergia on prenatal MIA offspring (Zolkipli-Cunningham et al. 2021).
A study by Naviaux et al. in 2013 investigated the effects of suramin on autism-like features in a mouse model(Naviaux et al. 2013). Researchers utilized the MIA mouse model, in which pregnant mice were exposed to poly(I:C) either once (E12.5) or twice (E12.5 and E17.5) to simulate viral infection. The resulting offspring exhibited ASD-like characteristics. Saline injected dams served as controls. Mice were treated with suramin at 6-weeks of age (10 or 20 mg/kg ip) and at 8-weeks animals were subjected to a series of tests. At 16-weeks, the males were sacrificed for analysis of cerebral synaptosomes and mitochondria, hematological tests, immunoglobulins, corticosterone, neuropathy and immunohistochemistry. Suramin treatment improved social preference, sensitometer coordination, metabolic rate/oxygen consumption and cerebral mitochondrial activity while normalizing body temperature. Neurobiological studies showed that this anti-purinergic agent prevented cerebellar Purkinje cell loss and corrected the synaptic structural abnormalities. The expression of purinergic receptors (P2Y2 and P2X7) and levels of Fmrp, which were reduced in the MIA mice, were also restored by suramin. Finally, suramin treatment corrected metabolic dysfunctions, increased cholinergic signaling in cerebral synaptosomes via the receptor nicotinic acetylcholine receptor subunit a7 (nAchRa7) and normalized signaling pathways involving ERK1/2 and CAMKII. This study underlines that aberrant purinergic signaling plays a crucial role in the development of ASD-like features and illustrates how anti-purinergic therapy with suramin can reverse these abnormalities in a mouse model. These findings provide a basis for further research into purinergic signaling as a potential therapeutic target for ASD.
In the following year, Naviaux et al. investigated the efficacy of a single dose of suramin to ameliorate the behavioral and metabolic abnormalities in the MIA-mouse model of autism (Naviaux et al. 2014). Male mice born to MIA-pregnant dams [administered two i.p. injections of Poly(I:C)] were treated with a single dose (20 mg/kg, i.p.) of suramin at 5.25-months of age, which is the biological equivalent of 30 years in humans. Two-days post-treatment, animals were evaluated for behavioral tests for novelty preference, social interaction, sensorimotor coordination, anxiety and light-avoidance. Five weeks after the suramin dose, half of the animals were sacrificed and their tissue levels of suramin were quantitated. The other half was again injected with the suramin at 7.5 weeks and sacrificed two days later to detect acute tissue levels of the suramin. Additionally, untargeted metabolic profiling was performed by mass spectrometry to determine the plasma metabolite levels of the animals that were screened for behavioral analysis. The effects of suramin peaked on day 2 and largely faded after 5 days, coinciding with its half-life. Metabolic pathways that were normalized were purine metabolism (ATP, ADP, AMP and adenosine levels) a key cell signaling pathway hypothesized to drive ASD-like symptoms, lipid metabolism which is crucial for brain signaling, and redox metabolism (balance of NADH/NAD +, glutathione systems) frequently associated with autism related oxidative stress, mitochondrial metabolism also known to be affected in autism, amino acid metabolism (glutamate, GABA) crucial for neurotransmission and excitatory and inhibitory balance, tryptophan metabolism which regulates mood and behavioral changes in ASD and others including glucose, nucleotide and carbon metabolism. Notably, two doses of suramin given at 6.5 and 6.75 months of age did not improve performance. This important
Comments (0)