Animals
The use of human adipose tissue was approved by the Ethics Committee of Gunma University (Approval ID: HS2021-150), and written informed consent was obtained from all donors prior to tissue collection. All animal experimental protocols were approved by the Gunma University Committee on Animal Care and Experimentation (Approval ID: 21–028) and were conducted in accordance with established institutional guidelines and the National Institutes of Health Guide for the Care and Use of Laboratory Animals [11]. BALB/c-nu/ + mice, sourced from the Shizuoka Laboratory Animals Center, Inc., Shizuoka, Japan, were selected for the study. Experiments were conducted exclusively on six-week-old female mice to maintain consistency across trials. The mice were housed at the Gunma University Institute of Experimental Animal Research, which maintains a specific pathogen-free environment to minimize external variables.
Adipose-derived stem cells
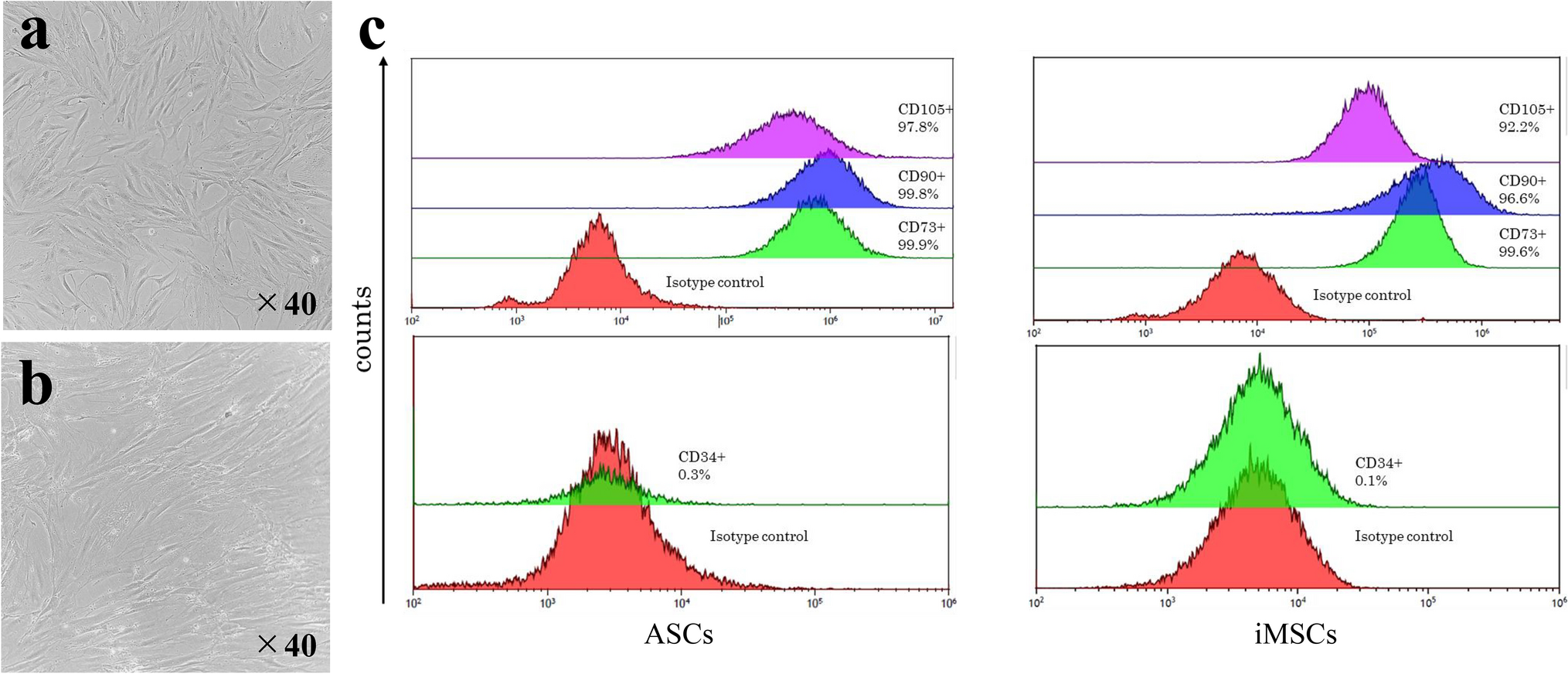
Human ASCs were isolated from excess subcutaneous adipose tissue obtained during breast reconstruction with deep inferior epigastric perforator (DIEP) flaps at the Department of Plastic and Reconstructive Surgery, Gunma University. The donors were breast cancer patients (n = 6; age range, 47–59 years); three had received systemic chemotherapy, three had received endocrine therapy, and none had received radiotherapy prior to tissue harvest. The ASC isolation protocol followed that described by Zuk et al. and Kakudo et al. [12,13,14]. Briefly, the excised adipose tissue was thoroughly washed with phosphate-buffered saline (PBS) and finely minced using sterile scissors. This tissue was then enzymatically digested with 1 mg/ml collagenase type II (Sigma-Aldrich C6885, St. Louis, MO, USA) in PBS, mixed in equal parts, and incubated with agitation for 40 min at 40 °C in a 50 cm3 centrifuge tube. The enzymatic reaction was quenched by adding 10 ml of Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Following centrifugation at 1600 rpm for 3 min, the floating adipose fraction and supernatant were discarded. The resultant cell suspension was then filtered through a 100-µm cell strainer (Corning, NY, USA) to remove debris and centrifuged again under the same conditions. The supernatant was aspirated, the pellet resuspended in an adequate volume of DMEM with 10% FBS, and the cell concentration determined. Cells were cultured in a humidified incubator at 37 °C with a 5% CO2 atmosphere. ASCs at passages three to five were utilized for the experiments to ensure optimal cell vigor and uniformity.
Induced mesenchymal stem/stromal cells
iMSCs were provided by the Center for iPS Cell Research and Application, Kyoto University. Upon receipt, the frozen iMSCs were promptly thawed and subsequently cultured to ensure their viability and functionality. Cultivation of these cells was performed using PRIME-XV MSC XSFM MDF1 medium (FUJIFILM Irvine Scientific, Santa Ana, CA, USA), a specialized formulation designed to support the growth and maintenance of MSCs under xeno-free conditions. The cultured cells were maintained in a humidified incubator at 37 °C with a 5% CO2 atmosphere. For the experiments, only cells that had undergone three to five passages were selected to ensure consistency in cell quality and to minimize variations that could affect the reproducibility of the results.
Flow cytometric analysis
Flow cytometry was employed to characterize the phenotypic profiles of isolated ASCs and iMSCs. A minimum of 1 × 10⁶ cells were suspended in 1.0 mL of DMEM supplemented with 1% FBS. The cell suspension was incubated with fluorochrome-conjugated monoclonal antibodies specific for CD90-PE, CD105-PE, and CD34-FITC (Beckman Coulter, USA). After staining, the labeled cells were analyzed using a Navios EX flow cytometer (Beckman Coulter, USA). After staining, the labeled cells were analyzed using a Navios EX flow cytometer (Beckman Coulter, USA). This analysis enabled the confirmation of mesenchymal stem cell marker expression and the exclusion of hematopoietic lineage contamination.
Flow cytometry was employed to characterize the phenotypic profiles of isolated ASCs and iMSCs. A minimum of 1 × 10⁶ cells were suspended in 1.0 mL of DMEM supplemented with 1% FBS. The cell suspension was incubated with fluorochrome-conjugated monoclonal antibodies specific for CD105-PE, CD90-PE, CD73-PE, and CD34-FITC (Beckman Coulter, USA). After staining, the labeled cells were analyzed using a Navios EX flow cytometer (Beckman Coulter, USA). This analysis enabled the confirmation of mesenchymal stem cell marker expression and the exclusion of hematopoietic lineage contamination.
Animal model
The fat grafting procedure used in this study was based on the Coleman technique [15], a recognized method in the field. For the purpose of the study, 0.3 ml of human adipose tissue was harvested using the liquid overflow method from surgically obtained excess fat. The harvested adipose tissue was meticulously minced using sterile scissors and subsequently introduced into a 2.5-ml syringe and combined with a specific cell solution prepared for each experimental group: 150 µl of PBS (control group, Group A); 150 µl of PBS containing 1 × 105 ASCs (Group B); and 150 µl of PBS containing 1 × 105 iMSCs (Group C). A total of sixteen cell solution preparations were created for each group to ensure consistency across experiments. Adipose tissue transplantation involved subcutaneous injection of the adipose tissue-cell solution mixture into two distinct sites on the dorsal surface of the animal model, utilizing an 18G needle to facilitate the procedure. Following transplantation, the grafted fat sites were monitored over a period of 28 days to assess the viability and integration of the grafts within the host tissue. Comprehensive analysis was performed on the extracted adipose tissue at the end of the observation period, on the 28th day, to evaluate the outcomes of the fat grafting procedure and the effects of the administered cell solutions.
Measurement of adipose tissue size
The dimensions of the transplanted adipose tissue were quantified in square millimeters (mm2) with the width-by-length method using calipers, as previously described [16]. To monitor the progression of tissue integration and growth, measurements were systematically taken at four-day intervals. This assessment regimen resulted in a total of eight measurements spanning the duration of the study, culminating on the 28th day post-transplantation.
Morphological assessment of adipose tissue
To evaluate the morphological characteristics of adipocytes within the three types of transplanted adipose tissue, samples were initially fixed in 10% formalin and subsequently embedded in paraffin. Thin sections of the embedded tissues were prepared and subjected to hematoxylin and eosin (H&E) staining to highlight cellular and tissue structures. These sections were then examined using light microscopy to assess adipocyte morphology. For a comprehensive analysis, sixteen fields at 100 × magnification were randomly selected across four H&E-stained slides for each adipose tissue type. Within these fields, the sizes of ten adjacent adipocytes were measured to determine the average adipocyte size in square micrometers (µm2). This process involved digital photography of each selected field, followed by precise measurement of adipocyte sizes on the photographs using ImageJ software (v.1.54, National Institutes of Health, Bethesda, MD), in alignment with protocols outlined in prior research [17]. This methodology enabled quantitative assessment of adipocyte size variations among the different transplant types, facilitating a detailed understanding of their morphological characteristics.
Assessment of fibrosis in transplanted adipose tissue
To quantify the extent of fibrosis within the transplanted adipose tissues, sections were subjected to Azan Mallory staining. Following fixation in 10% formalin and paraffin embedding, tissue sections were cut to a thickness of 4 µm. These sections then underwent a comprehensive deparaffinization process before being subjected to a series of staining steps designed to accentuate fibrotic regions. Specifically, the protocol involved incubating the specimens in Azocarmine G-drifting solution for 60 min, followed by treatment with 5% aqueous phosphorus molybdate. Subsequently, the sections were exposed to 30% hydrogen peroxide for 15 min and then stained with a mixture of Aniline blue and Orange G for 10 min. The staining process was completed using a series of dehydration, immersion, and occlusion steps to prepare the specimens for microscopic evaluation. For quantitative assessment of fibrosis, sixteen fields at 40 × magnification were randomly selected from four Azan Mallory-stained slides per adipose tissue type. The fibrotic area within each field was measured in square micrometers (µm2) using ImageJ software [18].
Assessment of neovascularization in adipose tissue
To evaluate neovascularization within the transplanted adipose tissues, capillary structures were identified through immunohistochemical staining using the von Willebrand factor, a marker that is widely used for highlighting endothelial cells in neovascular networks. The staining protocol started with an overnight incubation of adipose tissue sections at 4 °C with a rabbit polyclonal anti-von Willebrand Factor antibody (Abcam plc., Cambridge, UK). This initial step was followed by application of a biotinylated secondary antibody and then an HRP-conjugated anti-rabbit IgG goat polyclonal antibody (Nichirei Bioscience, Tokyo, Japan) to amplify the signal for antigenic detection. Visualization of antigen–antibody complexes was achieved using 3,3'-diaminobenzidine (DAB) as a chromogen, and the sections were subsequently counterstained with hematoxylin to enhance cellular detail. For quantitative analysis of neovascularization, sixteen fields at 40 × magnification were randomly selected from four immunostained slides corresponding to each type of transplanted adipose tissue. The area covered by neovascularization in each field was measured in square micrometers (µm2) using ImageJ software.
The fields for analysis were selected specifically from the graft margin—the transitional zone between the transplanted adipose tissue and host subcutaneous tissue—where angiogenic activity is typically highest. This region was defined histologically by the interface showing infiltration of host cells and new capillary structures, and was chosen to evaluate the contribution of MSCs to vascular ingrowth at the graft-host interface.
Histological analysis
Immunofluorescence staining was conducted to evaluate the presence and differentiation of transplanted stem cells within the adipose tissue. Paraffin-embedded sections were deparaffinized, rehydrated, and subjected to antigen retrieval by microwaving in 10 mM citrate buffer (pH 6.0). After blocking with 3% bovine serum albumin (BSA), sections were incubated overnight at 4 °C with the following primary antibodies: anti‑vimentin antibody (clone V9, catalog no. VIM‑V9‑L, Leica Biosystems, Germany), which recognizes human-specific vimentin, and anti‑perilipin A antibody (clone DBM15.6, catalog no. MOB534 [concentrated] or PDM534 [prediluted], Diagnostic BioSystems, USA), a marker of mature adipocytes. Fluorescently labeled secondary antibodies (Opal520 and Opal570; Akoya Biosciences, USA) were applied, followed by nuclear counterstaining with DAPI. Vimentin served as a marker for human mesenchymal stem cells (green), while perilipin indicated mature adipocytes (red). Quantitative analysis was performed using ImageJ software. The number of cells positive for both vimentin and perilipin (double-positive cells) was expressed as a percentage of the total number of perilipin-positive cells in each image field. Prior to detailed histological evaluation, low-magnification images were examined to identify structural zones within the grafted fat: peripheral capsule, viable fat tissue, and necrotic or fibrotic core. All subsequent analyses were limited to regions rich in viable adipocytes, carefully avoiding areas with apparent necrosis or dense fibrosis. Region-of-interest (ROI) selection was standardized across samples to minimize observer bias.
Statistical analysis
Statistical significance was evaluated using one-way analysis of variance (ANOVA), followed by a Tukey post hoc test, as deemed appropriate for comparison of groups. Prior to performing one-way ANOVA, data were tested for normality using the Shapiro–Wilk test and for homogeneity of variances using Levene’s test. Standard errors of the mean are depicted by error bars in graphical representations, with the number of experiments (n) stated for each experimental condition. Data were analyzed using EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan), a graphical user interface designed for R (The R Foundation for Statistical Computing, Vienna, Austria, v.4.1.2). EZR extends the capabilities of R Commander (v.2.7–1) by incorporating additional statistical functions that are commonly used in biostatistical analysis [19].

Comments (0)