Chemicals
The ability of the microbes to reduce PGE2 to PGF2α was tested using PGE2 kindly provided by Kyowa Pharma Chemical Co., Ltd. (Toyama, Japan). Arachidonic acid was purchased from Nu-Chek Prep, Inc. (Elysian, MN, USA). PGF2α, PGE2, PGD2, 11β-PGE2, 9α,11β-PGF2α, PGF2β (9β,11α-PGF2α), PGF1α, PGE1, PGF1β (9β,11α-PGF1α), PGF3α, and PGE3, which were used as standard samples, were purchased from Cayman Chemical (Ann Arbor, MI, USA).
Screening of a dried microbial cell library for PGE2 reduction
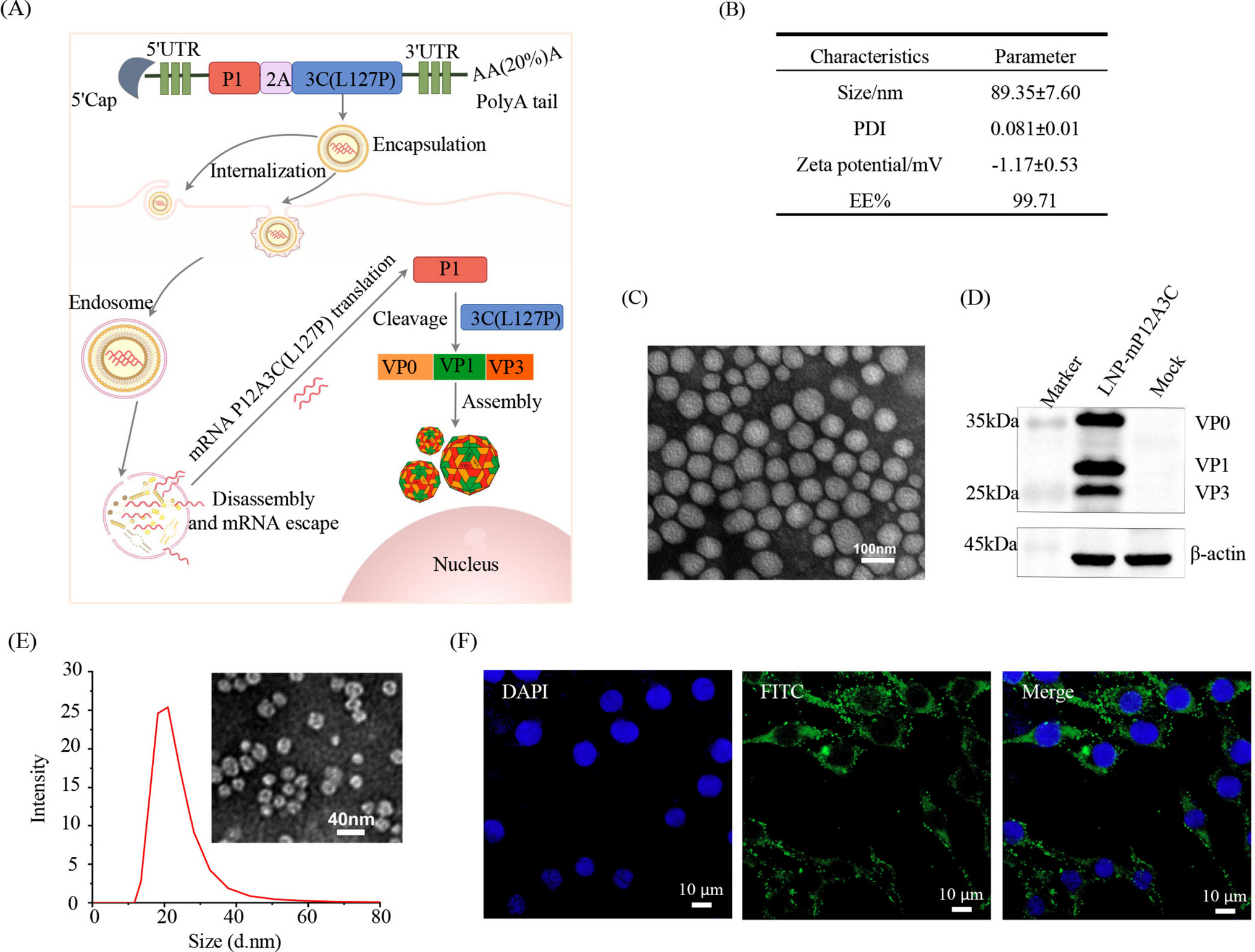
A dried microbial cell library from the AKU culture collection (Faculty of Agriculture, Kyoto University) was screened in this study (Table S1). One micro-spatula of the dried cells was placed into 10 mm test tubes, and 500 μL of the following reaction premix was added. The reaction premix contained 2.8 mM PGE2, 160 mM glucose, 3 mM NADH, 3 mM NADPH, and 14 U/ml glucose dehydrogenase (GDH, Amano Enzyme, Aichi, Japan) in 100 mM Tris–HCl (pH 8.0). The reaction was performed at 20 °C for 24 h with shaking at 300 rpm in a BR-23FP shaker (TAITEC, Saitama, Japan). After the reaction, substrate depletion and product formation were monitored using high-performance liquid chromatography (HPLC).
Microbial strains and culture conditions
R. kratochvilovae NBRC 0389 (AKU 4826) was inoculated into 5 mL of medium [5% (w/v) glucose, 0.5% peptone, 0.2% KH2PO4, 0.1% K2HPO4, 0.02% MgSO4∙7H2O, and 0.1% yeast extract, pH 6.5] and cultivated at 28 °C with shaking at 300 rpm for 18 h. The entire culture broth was added to 500 mL of the fresh medium, and the inoculated medium was cultivated at 28 °C with shaking at 120 rpm overnight. After cultivation, the cells were harvested by centrifugation at 6,000 × g for 20 min and washed twice with 0.85% (w/v) NaCl solution.
E. coli was cultivated in LB medium, which consisted of 1.0% (w/v) Bacto tryptone, 0.5% (w/v) Bacto yeast extract, and 1.0% (w/v) NaCl (pH 7.0). When necessary, ampicillin was added to the medium in a final concentration of 50 µg/mL.
Lipid extraction
Lipids in 500 μL of the reaction mixture were extracted using 500 μL of ethyl acetate after acidification with 10 μL of 1 M HCl. The mixture was centrifuged to separate the organic phase from the aqueous phase. The organic layer was transferred to a new tube, and the solvent was removed using a rotary evaporator. The extracted lipids were dissolved in 50 μL of ethanol and analyzed by HPLC.
Enzyme purification from R. kratochvilovae NBRC 0389
The harvested cells from 500 mL of culture medium were suspended in 20 mM [4-(2-hydroxyethyl)−1-piperazineethanesulfonic acid] (HEPES, pH 8.0) containing cOmplete (Sigma-Aldrich; St. Louis, MO, United States) at a buffer-to-cell weight ratio of 2:1. The cell suspension was mixed with an equal weight of glass beads (0.5 mm diameter), and the cells were disrupted using a Multi-beads shocker (Yasui Kikai Corp, Osaka, Japan) with 6 cycles of 60 s on/off at 2500 rpm. Cell-free extracts (CFE) were obtained by centrifugation (12,000 × g, 20 min). The CFE was separated by ultracentrifugation (100,000 × g, 60 min), and the supernatant was collected. The ultracentrifugation supernatant was desalted and concentrated to 10 mL using a VivaSpin Turbo 50 (10,000 MWCO; Sartorius, Goettingen, Germany).
The ultracentrifugation supernatant (10 mL) was applied to an ÄKTA explorer fast protein liquid chromatography system (Cytiva, Tokyo, Japan) equipped with a HiPrep Diethylaminoethyl Sepharose Fast Flow (DEAE FF) 16/10 (10 mL) column (Cytiva) equilibrated with 20 mM HEPES (pH 8.0). After the column was washed with 5 column volumes of buffer, linear gradient elution was performed with 20 mM HEPES (pH 8.0) containing 1 M NaCl for 10 column volumes at a flow rate of 1 mL/min. These fractions were evaluated for PGE2-reducing activity, and the active fractions were collected. The active fractions were desalted and concentrated using a VivaSpin Turbo 15 (10,000 MWCO; Sartorius). The active fraction from the DEAE FF 16/10 step was applied to a Mono Q 10/100 GL column (Cytiva) equilibrated with 20 mM HEPES (pH 8.0). After washing with 5 column volumes of buffer, linear gradient elution was performed with 20 mM HEPES (pH 8.0) containing 1 M NaCl for 10 column volumes at a flow rate of 2 mL/min. Fractions were evaluated for PGE2-reducing activity, and the active fraction was collected.
Protein analysis
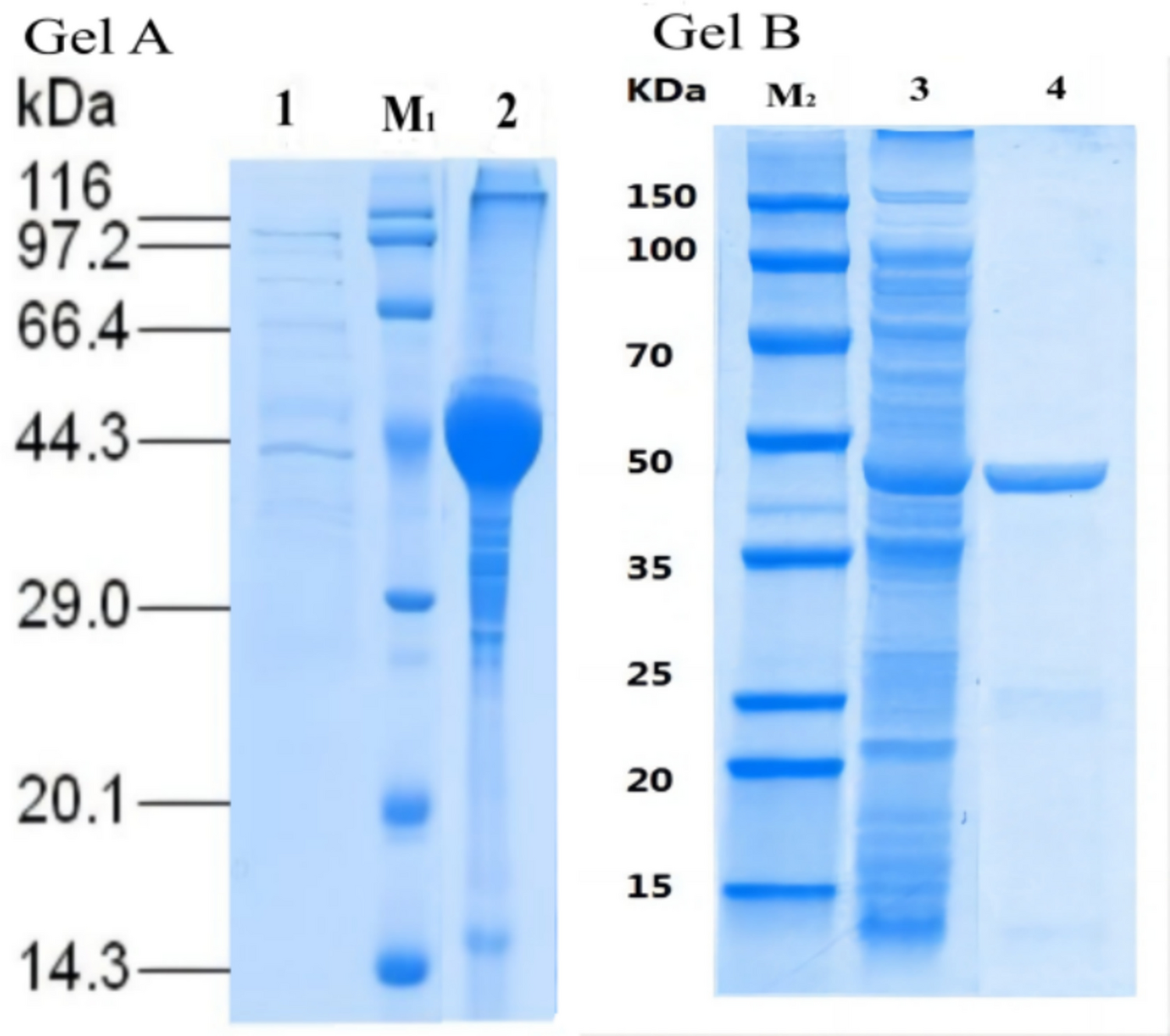
The protein concentration was determined using the Quick Start Bradford protein assay Kit (Bio-Rad Laboratories, Inc.). The protein profiles at each purification step were analyzed using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) using 10% polyacrylamide gels.
Determination of the N-terminal amino acid sequences
The proteins separated by the SDS-PAGE were electroblotted onto a polyvinylidene fluoride or polyvinylidene difluoride (PVDF) membrane to analyze their amino acid sequences. The N-terminal amino acid sequence of each excised protein band was determined by automated Edman degradation using a PPSQ-31A protein sequencer (Shimadzu, Kyoto, Japan).
Cloning of prostaglandin F2α 9-dehydrogenase (PDH) encoding gene
Total RNA from R. kratochvilovae NBRC 0389 was extracted using the RNeasy® Plant Mini Kit (QIAGEN, Venlo, The Netherlands) according to the manufacturer’s instructions. Next-generation sequencing (150 bp paired-end) on the DNBSEQ-G400 and de novo assembly were performed at the Bioengineering Lab. Co., Ltd. (Kanagawa, Japan). An open reading frame was found in the nucleotide sequence, and it was predicted to encode a PDH enzyme containing an N-terminal amino acid sequence corresponding to one of the purified proteins. The PDH gene and a malate dehydrogenase (MDH) candidate gene were codon-optimized for E. coli, synthesized (Eurofins Genomics Co., Tokyo, Japan), and amplified by polymerase chain reaction (PCR) using a Biosystems® ProFlex™ PCR System (Thermo Fisher Scientific, Waltham, MA, USA) using the following primers. To amplify the PDH gene, sense primer 5′-GTTTAACTTTAAGAAGGAGATATACATATGGCCCCTTTCACCTACCT-3′ and antisense primer 5′-CGGATCTCAGTGGTGGTGGTGGTGGTGCTCCCACGGCATGGTTTC-3′ (underlined sequences indicate the vector region) were used. To amplify the MDH gene, sense primer 5′-GTTTAACTTTAAGAAGGAGATATACATATGGGCCTGAAAACCGCA-3′ and antisense 5′-CGGATCTCAGTGGTGGTGGTGGTGGTGCTCTTACAGCTTGGAACCCTGG-3′ were used. PrimeSTAR GXL DNA Polymerase (Takara Bio Inc., Shiga, Japan) was used as a DNA polymerase, and the reaction composition was in accordance with the manufacturer’s instructions. Amplification was performed using 30 cycles of 98 °C for 10 s, 60 °C for 15 s, and 68 °C for 1 min. The PCR product was cloned into the vector pET21-b, digested with NdeI and XhoI using the In-Fusion HD Cloning Kit (Takara Bio Inc.). The resulting plasmid, pET-21b/His-tag-PDH, was introduced into E. coli BL21 (DE3) (Merck, Darmstadt, Germany) by heat shock.
Accession number(s)
The cDNA sequences of R. kratochvilovae NBRC 0389 were deposited in the International Nucleotide Sequence Databases (INSD) under accession numbers ICXD01000001–ICXD01040545. The PDH gene and the MDH gene from R. kratochvilovae NBRC 0389, modified for heterologous expression in E. coli, were deposited in INSD under accession numbers LC877254 and LC905278.
Expression and purification of recombinant PDH
E. coli BL 21(DE3) harboring pET-21b/His-tag-PDH was induced, collected, and disrupted by the same method of our previous paper (Kimoto et al. 2022). PDH was purified from the cell free extracts by affinity chromatography with a HisTrap HP (5 mL; Cytiva) using the same method of our previous paper (Kimoto et al. 2022).. Finally, the PDH solution was mixed with an equal volume of glycerol and stored at − 20 °C until use. All the procedures were performed at 4 °C.
Enzymatic characterization of the PDH by absorbance measurement
The PGE2-reducing reaction was performed in a 200 μL reaction mixture containing 20 mM phosphate buffer (pH 8.0), 5 mM PGE2, 0.2 mM NADPH, and 1.2 µM (31 mg/L; 4.3 U/L) PDH. PGF2α-dehydrogenating reaction was performed in a 200 μL reaction mixture containing 20 mM phosphate buffer (pH 8), 5 mM PGF2α, 5 mM NADP+, and 1.2 µM (31 mg/L; 4.3 U/L) PDH. The absorbance of NADPH consumed in the PGE2 reduction or generated in the PGF2α dehydrogenation was measured over time at 340 nm using a UV–visible spectrometer UV-1800 (Shimadzu, Kyoto, Japan) or a microplate reader SpectraMax250 (Molecular Devices, San Jose, United States). The optical path length of the instruments was 1 cm and 0.625 cm, respectively.
Effects of temperature and pH on the reactions
To determine the optimal reaction temperature and pH of PDH, enzyme assays were conducted at various temperatures from 18 °C to 48 °C in increments of 5 °C and with buffers of different pH values, i.e., 20 mM KPB (pH 6.0, 6.5, 7.0, 7.5, and 8.0), Tris–HCl buffer (pH 8.5 and 9.0), and sodium bicarbonate buffer (pH 9.5, 10.0, and 10.5). To evaluate thermal stability, the enzyme solution was pre-incubated for 15 min at 4 °C (on ice) or at temperatures from 0 °C to 50 °C in increments of 10 °C with 20 mM KPB (pH 8.0). Enzyme activities were assayed at 28 °C (except for optimal temperature determination) in 20 mM Tris–HCl buffer (pH 8.0) (except for the optimal reaction pH). The enzyme activities were evaluated by measuring the NADPH concentration via the absorbance at 340 nm.
Substrate specificity of PDH
Five kinds of substrates, PGE1, PGE2, PGD2, 11β-PGE2, and PGE3, were tested as substrates for the reduction by the PDH. The reduction reaction was performed in 20 mM KPB (pH 8.0) containing 5 mM substrate, 7.2 µM (186 mg/L;26 U/L) PDH, 100 mM glucose, 15 U/mL GDH, and 2 mM NADPH. Additionally, six substrates (PGF1α, PGF1β, 9β-PGF2α, PGF2α, 9α,11β-PGF2α, and PGF3α) were tested for dehydrogenation. The dehydrogenation reaction was performed in 20 mM KPB (pH 8.0) containing 5 mM each substrate, 4.8 µM (124 mg/L;17 U/L) PDH, and 5 mM NADP+. Reactions were performed at 28 °C for 18 h with shaking at 200 rpm. The reaction products were extracted as described and analyzed by HPLC and Liquid Chromatography–Mass Spectrometry (LC–MS).
HPLC and LC–MS analyses
Quantitative analysis of PGs was performed by HPLC using a Prominence HPLC system (Shimadzu) equipped with a COSMOSIL 5C18-AR-II Packed Column (4.6 mm I.D. × 150 mm, Nacalai Tesque, Kyoto, Japan) at 40 °C. Analysis and LC–MS conditions were the same as described in our previous paper (Fujii et al. 2025).
Non-stereospecific chemical reduction of PGE2 and PGD2 by sodium borohydride
To prepare PGF2α diastereomers, the keto groups of PGE2 and PGD2 were reduced chemically without stereospecificity. Fifty micrograms of either PGE2 or PGD2 was placed into a test tube. Subsequently, 4 μL of 0.5 M cerium (III) chloride in methanol, 2.5 μL of 0.5 M sodium borohydride (NaBH4) in methanol, and 13 μL of water were added into the tube in that order. After acidification with 1.3 μL of 1 M HCl, lipids were extracted from the reaction mixture with 50 μL of ethyl acetate.


Comments (0)