
Remember me
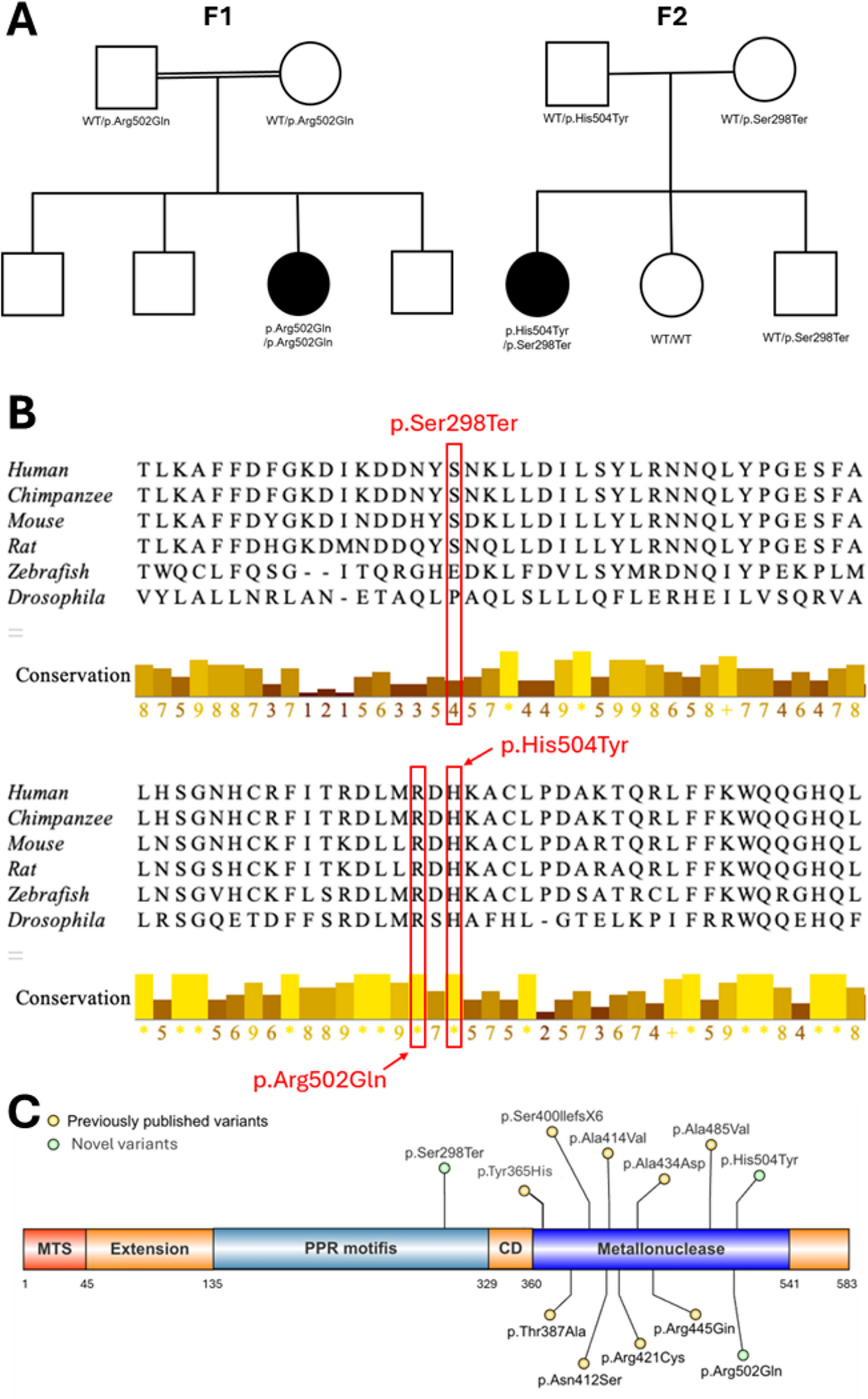
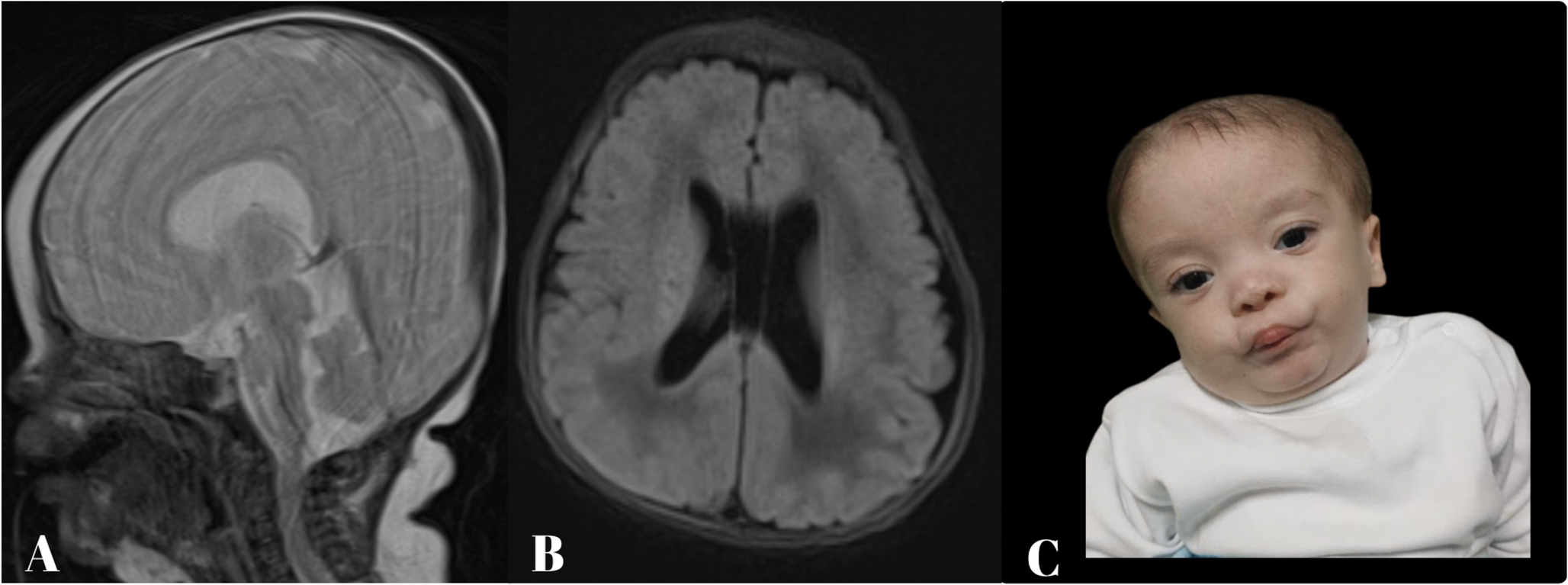
Family F1 is consanguineous (distant relatives) and originates from Upper Egypt. The proband is female aged 5 years and 2 months, born to healthy parents, with three unaffected male siblings. No similar conditions were reported in the family. She presented with global developmental delay, growth retardation, and SNHL. Motor milestones were delayed; head control was achieved at 1 year, and at her most recent evaluation at 3 years, she was only able to sit with support. Cognitive function was severely impaired, with absent speech, limited environmental recognition, and a lack of sphincter control. There was no history of seizures. On physical examination, her weight was 10 kg (-4.1SD), height was 90 cm (-3.9SD), and head circumference was 47.5 cm (-2.1SD). Dysmorphic facial features were noted, including a triangular face with an open mouth, high forehead, arched eyebrows, broad nasal root and bridge, bulbous nose, long philtrum, everted lower lip, and low-set ears. Neurological examination revealed generalized hypotonia with preserved deep tendon reflexes. Investigations, including karyotyping, extended metabolic screening, urinary organic acids, thyroid profile, electromyography, nerve conduction studies, and echocardiography were unremarkable. Serum lactate was elevated at 27 mg/dL (reference 4.5–19.8), and ammonia was 68 mg/dL (reference 15–45). Auditory brainstem responses demonstrated bilateral severe-to-profound hearing loss. Brain MRI was normal, except for a thin corpus callosum and the presence of a cavum septum pellucidum. Exome sequencing identified a homozygous missense variant in PRORP (NM_014672.4), c.1505G > A (p.Arg502Gln). Both parents were healthy heterozygous carriers.
The proband from family F2 is a 12-year-old female of Honduran ancestry born to non-consanguineous parents who have two other clinically unaffected children. She was born at full term. She walked at 2 years and used small phrases at 2-2.5 years of age. However, there was clear regression (marked by the loss of ability to walk and talk) after she started to have multiple seizure types from around 2 years. These seizures were primarily bilateral tonic-clonic, but also tonic and hemiclonic. She has had multiple episodes of status epilepticus. Currently, she is nonverbal, non-ambulatory, has profound intellectual disability and her physical exam shows severe hypotonia and scoliosis. She has bilateral SNHL and oral dysphagia. Her brain MRI shows patchy encephalomalacia involving the right more than the left cerebral/cerebellar border zones (Figure S1-3). Seizure control has been challenging despite multiple medications. She has been seizure free for 2 months with a combination of levetiracetam and clobazam. No assessment of ovarian function has been undertaken. Trio exome sequencing of the proband revealed compound heterozygous variants in PRORP (NM_014672.4: c.1159 A > G, p.His504Tyr and c.893 C > A, p.Ser298Ter). No variants in other candidate genes or any of the known Perrault syndrome genes were identified. Both variants were confirmed by Sanger sequencing and segregation analysis, which reveled that her unaffected parents are heterozygous, whilst her younger brother was heterozygous for the nonsense variant and her younger sister is homozygous for the wild-type allele.
The p.Ser298Ter variant is predicted to result in a null allele secondary to nonsense mediated decay. The missense variants, p.Arg502Gln and p.His504Tyr, alter highly conserved residues among PRORP orthologues (Fig. 1B) and are predicted to be deleterious by multiple in silico analyses (Table S2). Within gnomADv4.1, these variants are found at frequencies consistent with an ultra-rare autosomal recessive disorder [7].
Fig. 1 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.PRORP family pedigrees, conservation analysis of variant residues and variant locations. (A) Pedigrees for families F1 and F2, each with biallelic PRORP variants in the proband. (B) Position of missense variant residues highlighted in red. Symbols below the sequence alignments represent level of conservation across species, conservation scores range from 0 (least conserved) to 11 (full conservation), indicated by * (asterisk), based on AMAS method. Alignment visualised using Jalview software. (C) Schematic illustrating the location of all established PRORP variants to date (NM_014672.4). Novel variants are coloured in green, whilst previously published variants are in yellow. Figure created using IBS 2.0 [8] MTS = mitochondrial targeting sequence. PPR = pentatricopeptide repeat. CD = central domain.
We considered the effect of the two missense changes, by generating variant protein models of PRORP in the context of the mtRNase P complex. In the reported structure (PDB 4XGL), residue Arg502 makes direct contact with the acceptor arm and leader nucleotides of the pre-tRNA substrate, positioning the scissile phosphodiester bond in the nuclease active site [9, 10] (Fig. 2). Mispositioning of the pre-tRNA substrate may partially destabilise the optimal conformation for metallonuclease activity. p.Arg502Gln removes a hydrogen bond to pre-tRNA-His(5,Ser), weakening specific RNA contacts. In contrast, residue His504 is located close to one of the metal-binding residues (Asp503), within the metal 2 catalytic pocket [9, 10]. The p.His504Tyr substitution introduces a bulkier tyrosine that fills a cavity and improves packing but increases the steric repulsion (fa_rep term + 2.8 REU), as well as introduces one additional buried, unsatisfied polar atom from the Tyr hydroxyl. ΔSASA and ΔΔG show negligible changes in both cases.
Fig. 2 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Structural analysis of PRORP variants in the context of the mtRNase P complex. PRORP variants p.Arg502Gln and p.His504Tyr modeled on the cryo-EM structure of RNase P in complex with mitochondrial pre-tRNA-His(5,Ser) (PDB 8CBK) using PyRosetta. Inset, left: p.Arg502Gln (violet side-chain) removes an RNA hydrogen bond. Inset, right: p.His504Tyr(green side-chain) fills a cavity and improves packing but increases steric repulsion and buries an unsatisfied hydroxyl. Variant side chains are shown as sticks; hydrogen bonds as dashed lines
To determine whether the PRORP missense variants disrupted mitochondrial tRNA processing, we purified recombinant PRORP proteins carrying the variants and assessed mtRNase P complex endonucleolytic activity of pre-tRNAIle [2]. mtRNase P complexes containing the p.Arg502Gln or p.His504Tyr variants significantly diminished 5’ cleavage product levels compared to wildtype PRORP (p < 0.0001; Fig. 3), with relative cleavage activity decreases of 33% and 61%, respectively.
Dermal fibroblasts were not available from either affected individual to undertake OXPHOS studies or measure the levels of PRORP.
Fig. 3 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Functional assessment of PRORP variants in mtRNase P tRNA processing assays. Cleavage of the pre-tRNAIle 5′ leader sequence by mtRNase P containing wild-type (WT) or variant PRORP. The intensity of the pre-tRNAIle cleavage product was quantified, and the variants were normalized against wild-type PRORP. Error bars represent the standard error of the mean. N = 5, ****p < 0.0001, one-way ANOVA with Dunnett’s multiple comparisons test, comparing wild-type to variants
Comments (0)