This report describes inflammatory cerebral amyloid angiopathy (CAA-RI) in a 32-year-old woman with Down syndrome (DS), an unusually early age of onset compared with the typical mean of 67 years [1]. While cerebral amyloid angiopathy (CAA) is common in DS due to amyloid precursor protein (APP) gene triplication [7], the inflammatory subtype is rare. Neuropathological and microglial data in DS further support a pro-inflammatory milieu around vascular amyloid. Published biomarker studies in DS (including sTREM2 and cytokine signatures) provide a plausible substrate for such susceptibility, even though the studies in our report are not patient-derived [8,9,10,11,12,13,14,15]. This framework explains the early onset (32 years) of CAA-RI despite the > 40-years age threshold in the general criteria and aligns with the radiological evolution in which susceptibility markers may be absent at onset and become apparent later.
The sequence in this patient, systemic inflammatory flare followed by subacute neurological deficits with asymmetric edema and leptomeningeal enhancement, raises a pathophysiological link in Down syndrome, where innate immune dysregulation lowers the threshold for CNS inflammation [12,13,14]. However, temporal association alone does not establish causality, and alternative or concurrent triggers cannot be excluded. The subsequent acute neurological deficits, including hemiparesis and seizures, align with common CAA-RI presentations such as cognitive decline and headache [1, 10,11,12].
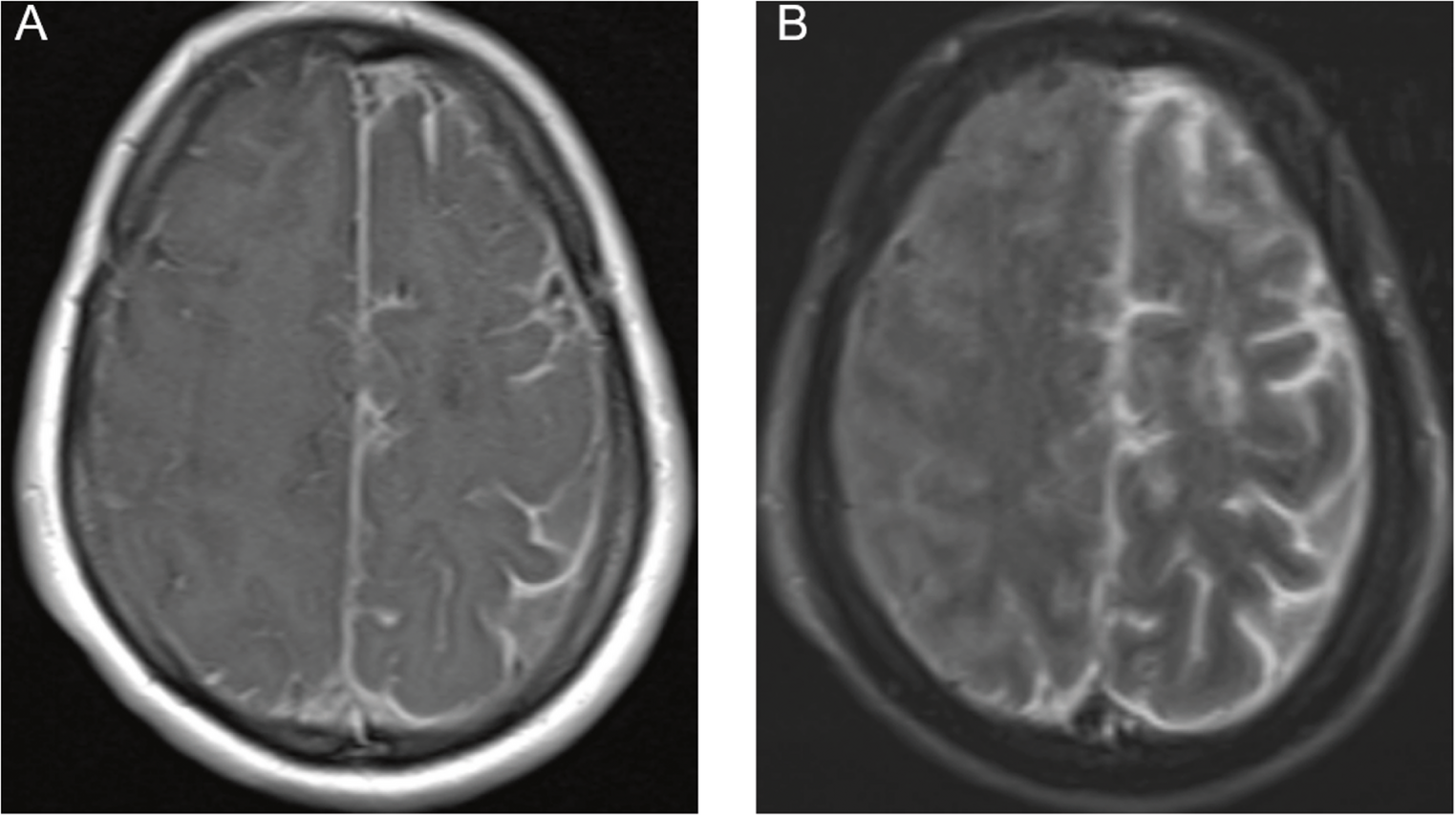
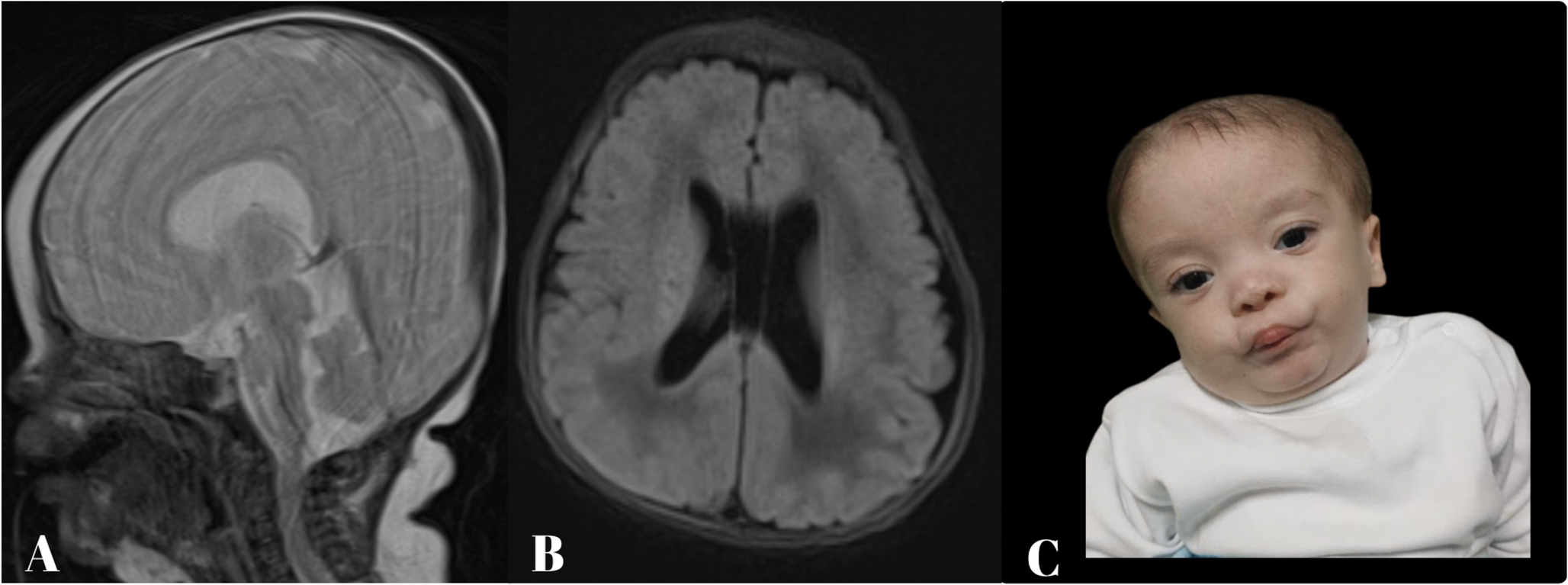
The diagnosis was supported by characteristic neuroimaging and cerebrospinal fluid (CSF) analysis. The patient met criteria for probable CAA-RI. Table 1 compares Auriel et al.‘s (2016) diagnostic criteria with our patient. Brain MRI disclosed asymmetric T2/FLAIR hyperintensities with leptomeningeal enhancement and focal superficial siderosis on susceptibility-weighted imaging, without microbleeds; cerebrospinal fluid showed marked inflammation. Although microbleeds are frequent markers of underlying CAA [1, 8, 16, 17], their absence does not exclude CAA-RI early in the disease, particularly when the clinicoradiological constellation is otherwise typical. The CSF showed marked inflammation, with high protein and pleocytosis, consistent with over 80% of CAA-RI cases [6, 18]. Although not performed here, the APOE ε4/ε4 genotype is a significant risk factor for CAA-RI, and CSF Aβ levels may be altered [3]. Anti-amyloid-β autoantibodies were not assessed in this patient.
Table 2 Diagnostic work‑upAlthough the Auriel criteria typically require age > 40 years for the diagnosis of CAA-RI, Down syndrome represents a well-recognized exception due to its accelerated amyloid pathology. Neuropathological studies have demonstrated that virtually all individuals with Down syndrome develop full Alzheimer’s disease pathology by the fourth decade, with cerebral amyloid angiopathy emerging as early as the third decade [2]. Microbleeds and vascular amyloid deposition appear in the mid-to-late thirties, significantly earlier than in the general population [19].
Our patient, aged 32 years, falls within this expected window for CAA-related complications in Down syndrome. The triplication of the APP gene on chromosome 21 results in lifelong amyloid-β overproduction [7], making Down syndrome one of the strongest genetic risk factors for early-onset CAA. A case of APP locus triplication with CAA onset at 39 years has been reported [2], only seven years older than our patient. These observations support adapting CAA-RI diagnostic criteria for Down syndrome, with age thresholds adjusted to account for the accelerated timeline of amyloid deposition characteristic of this population.
Brain biopsy was deferred given fulfillment of probable CAA-RI with high-specificity clinicoradiological criteria, typical MRI and inflammatory CSF, exhaustive negative infectious/neoplastic evaluations, and a rapid response to immunosuppression; biopsy was reserved for atypical evolution or treatment failure.
The treatment course reflects the variable response of CAA-RI to immunosuppression. High-dose corticosteroids are first-line therapy, and immunosuppressive treatment is associated with better clinical outcomes and fewer recurrences [6, 7, 20]. In this case of partial steroid response, rituximab was used, a strategy supported by observational data for maintenance therapy in refractory CAA-RI [21, 22]. Patients with the Aβ-related angiitis (ABRA) subtype may require combination therapy more often, though relapse rates can remain high compared to primary angiitis of the central nervous system (PACNS) [21].
The primary limitation is the lack of neuropathological confirmation. The diagnosis of probable CAA-RI was based on clinicoradiological criteria with high sensitivity and specificity [23], but a brain biopsy was not performed. As a single case report, these findings are not generalizable to the heterogeneous DS population.
This case shows that, in Down syndrome, accelerated vascular amyloid makes age < 40 an inadequate exclusion for CAA‑RI; when clinicoradiological criteria are met, CAA‑RI should be considered irrespective of age. It highlights a potential interaction between the early amyloid pathology in DS and the population’s unique neuroinflammatory state [15, 24]. Future prospective studies using advanced biomarkers are needed to better define the incidence, triggers, and optimal management of CAA-RI in this specific population.

Comments (0)