
Remember me
The patient was born to healthy parents without a family history of neurological disorders nor consanguinity. From approximately 6–7 years of age, her academic performance was slightly below average, and she had mild coordination difficulties in sports. However, these features were subtle and nonspecific, and their relevance to the later neurological disorder remains uncertain. After graduating high school, she began working in an office job.
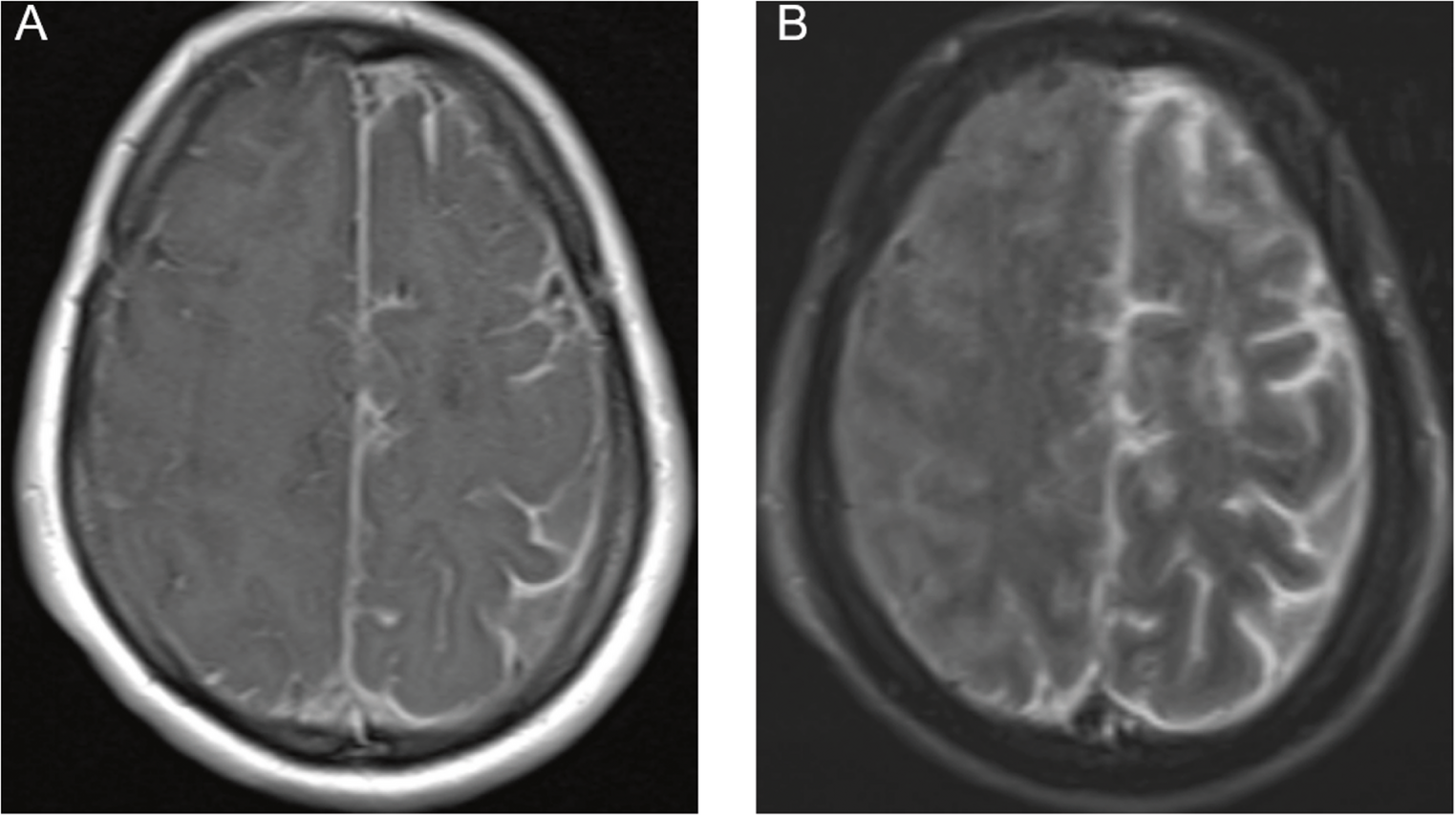
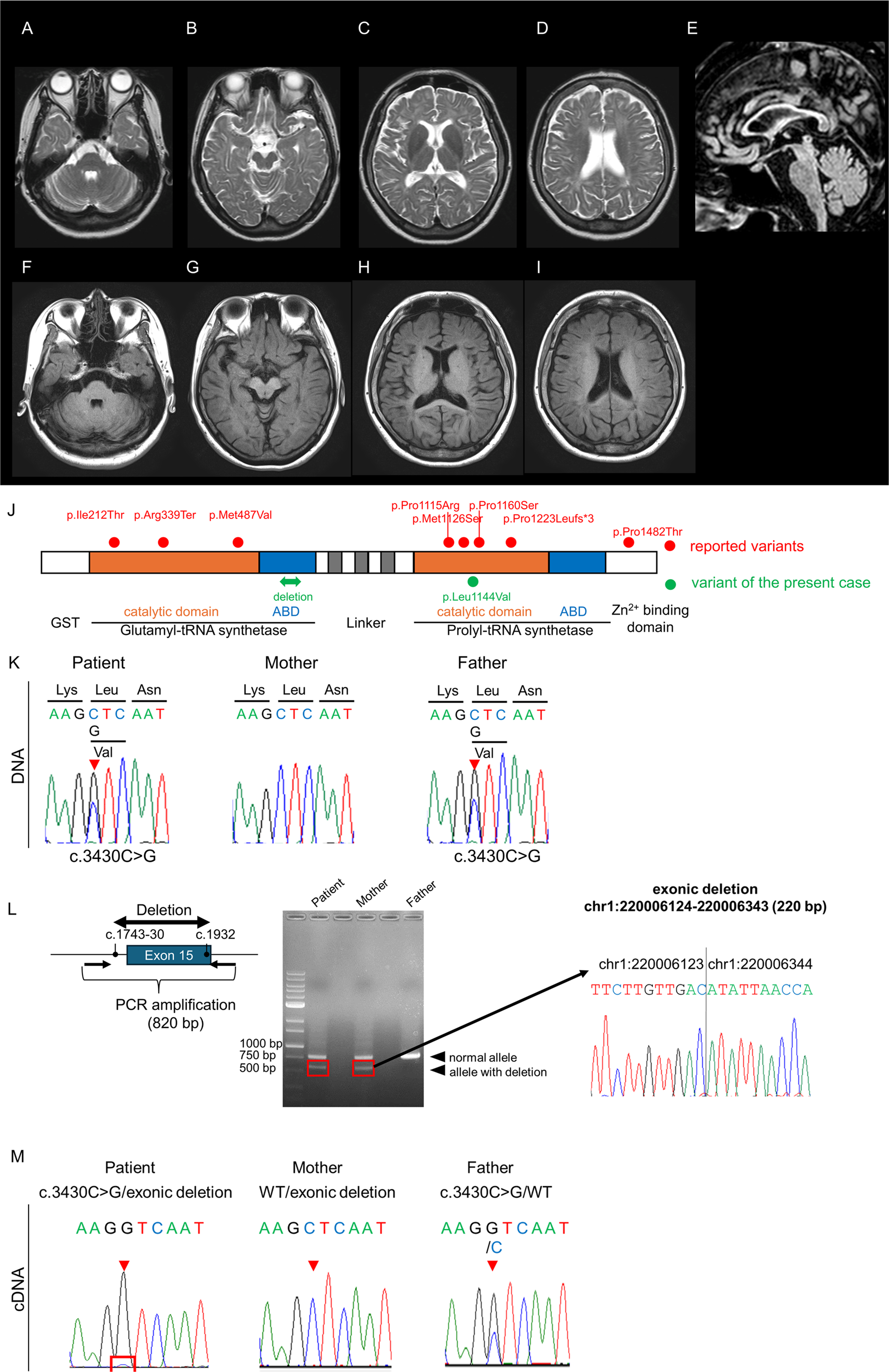
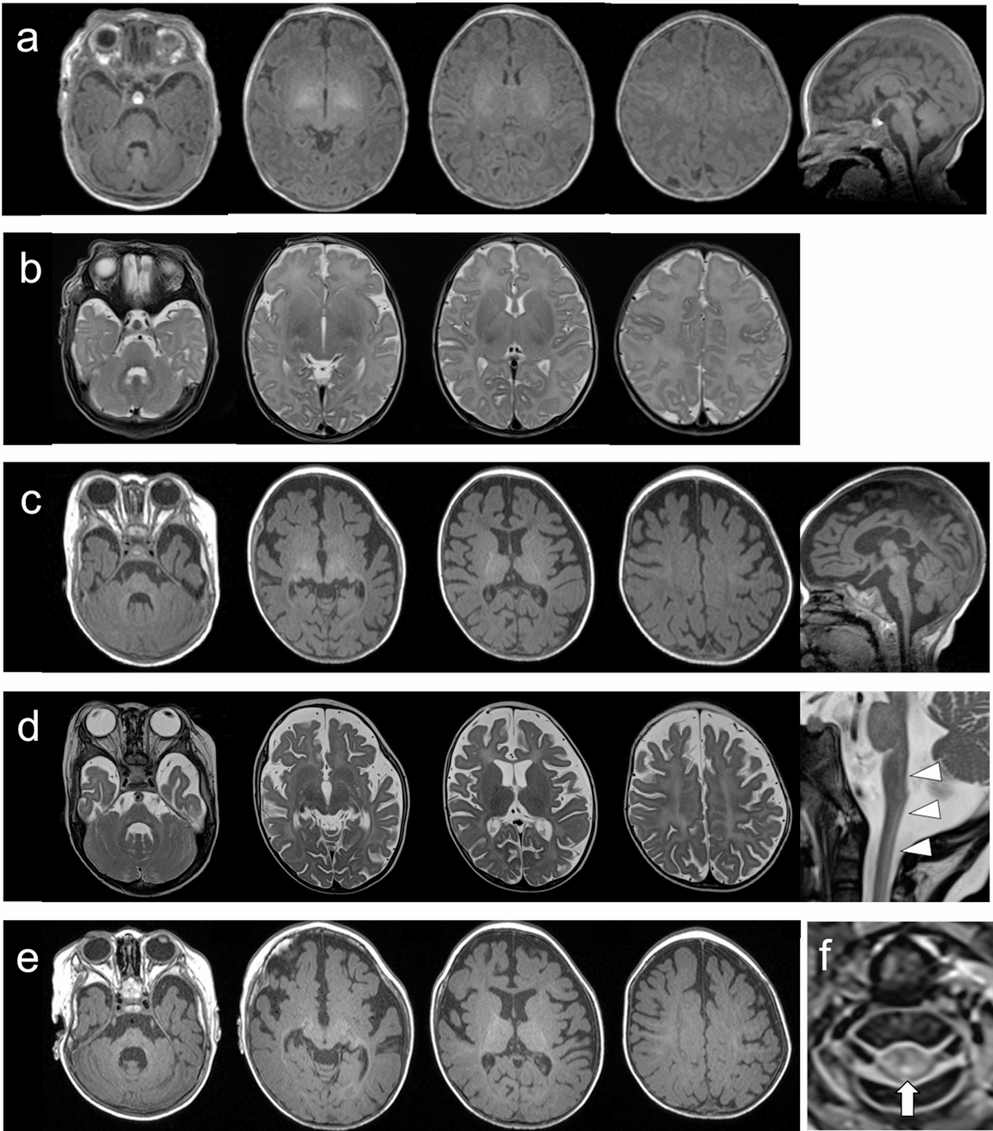
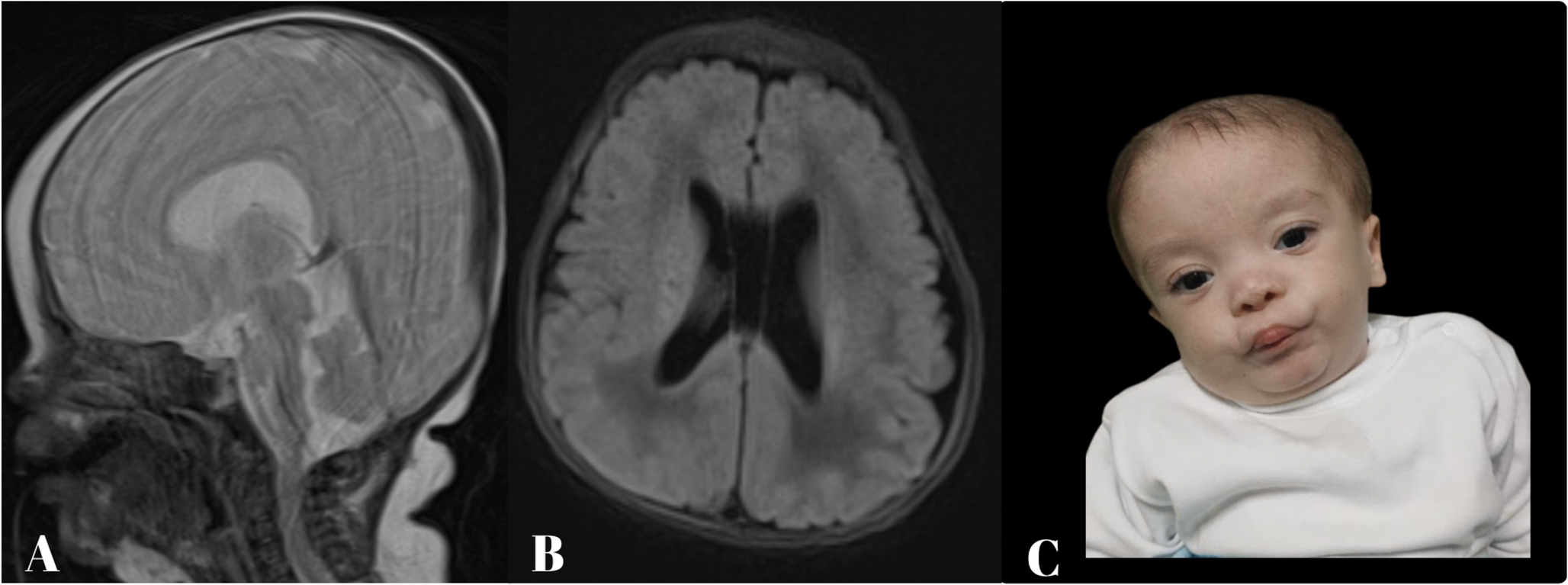
At the age of 32, she began experiencing difficulty with writing, accompanied by neck tremors. Her writing difficulties gradually worsened, and she later developed dysarthria and incoordination. By age 40, the patient showed worsening gait instability and tremor, which prompted her to visit an outpatient clinic. Brain MRI revealed extensive cerebral white matter atrophy accompanied by hyperintensity on T2-weighted images, with thinning of the corpus callosum (Fig. 1A–E). T1-weighted images showed slight hyperintensity of the periventricular white matter and basal ganglia relative to cortex (Fig. 1F–I). Leukoencephalopathy with unknown etiology was suspected, and she was referred to our hospital for further evaluation.
Fig. 1 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Brain MRI and genetic analysis of the present case. (A–D) Axial T2-weighted images obtained at age 40 show hyperintensity in the subcortical white matter. No abnormal signal intensity is observed in the middle cerebellar peduncle. (E) Sagittal T1-weighted image demonstrates thinning of the corpus callosum. (F–I) Axial T1-weighted images show slight hyperintensity of the periventricular white matter and basal ganglia relative to cortex. (J) Schematic representation of the EPRS1 gene indicating the positions of pathogenic variants within functional domains. ABD, anticodon-binding domain. (K) Sanger sequencing of EPRS1 reveals a heterozygous missense variant, c.3430 C > G (p.Leu1144Val), in the patient and her father. (L) Agarose gel electrophoresis of PCR products from genomic DNA using primer pairs flanking the deleted region shows that the patient and her mother share the deletion (left). The electropherogram of the breakpoint junctions indicates a 220-bp deleted region (right). (M) Electropherogram of RT-PCR products demonstrates monoallelic expression of the c.3430 C > G variant in the patient. The mother does not carry the variant, while the father is heterozygous for c.3430 C > G
Neurological examination revealed slurred speech, ataxia in the limbs and trunk, focal hand dystonia, and postural tremor in the neck. In cognitive assessments, the patient scored 26 out of 30 on the Mini-Mental State Examination, 23 of 30 on the Japanese version of the Montreal Cognitive Assessment, and 12 of 18 on the Frontal Assessment Battery, indicating mild cognitive impairment. HLD was suspected, and genetic analysis was performed.
The patient and her parents provided written informed consent. The study was approved by the institutional review board. Genomic DNA samples were prepared from peripheral blood leukocytes obtained from the patient and her parents.
We conducted Whole-exome sequencing (WES) of the patient using the SureSelect Human All Exon V6 + UTRs kit (Agilent Technology, Santa Clara, CA) with the HiSeq 2500 platform (Illumina, San Diego, CA). The sequences were aligned to the GRCh37/hg19 reference genome using the Burrows Wheeler Aligner, and variants were called with SAMtools [7, 8]. After initial variant filtering (quality score > 20 and minor allele frequency < 0.01) against our in-house exome database of 1,163 Japanese controls, 491 rare variants remained. We then prioritized variants in genes associated with hypomyelinating leukodystrophies and related neurodevelopmental disorders (Supplementary Table 1). This analysis yielded a single plausible candidate consistent with the phenotype: a heterozygous missense variant in EPRS1 (c.3430 C > G; p.Leu1144Val) (NM_004446.3). Trio-based analysis did not identify any plausible de novo variants consistent with the phenotype relevant to leukodystrophy.
This variant has not been registered in HGMD, ClinVar, gnomAD v4.1.0 (allele frequency = 0), or ToMMo 54KJPN and affects an amino acid conserved across vertebrates. It is predicted to be deleterious, with a Combined Annotation Dependent Depletion v1.6 Phred score of 26.6. The variant is located in the Prolyl-tRNA synthetase (ProRS) domain of EPRS1, a region where most pathogenic variants have been reported, including the pathogenic c.3377T > C (p.Met1126Thr) variant in the same exon (Fig. 1J). Segregation analysis confirmed that the missense variant was paternally inherited (Fig. 1K). However, no other SNVs or short indels were detected in EPRS1.
Whole-genome sequencing (WGS) was performed at the National Center for Global Health and Medicine using the NovaSeq 6000 platform (Illumina, San Diego, CA) with 150-bp paired-end reads at a target depth of 30× [9]. Reads were aligned to the GRCh38/hg38 reference genome, and variant calling was conducted using Parabricks v3.1.0 (NVIDIA, Santa Clara, CA), which accelerates GATK-recommended analyses through GPU processing [10].
Structural variant (SV) analysis was performed on WGS BAM files aligned to GRCh38. SVs were identified with the Manta structural variant caller [11]. Manta called a 220‑bp deletion in EPRS1 involving part of exon 15 [NC_000001.11:g.220006124_220006343del; NC_000001.11(NM_004446.3):c.1743-30_1932del]. To define the exact breakpoints, we designed PCR primers flanking the breakpoint junction. Direct sequencing of the PCR products confirmed the deletion [NC_000001.11:g.220006124_220006343del; NC_000001.11(NM_004446.3):c.1743-30_1932del], concordant with the Manta call. This SV was not registered in gnomAD SVs v4.1.0 (allele frequency = 0). The exon 15 deletion was maternally inherited (Fig. 1L). The paternally inherited missense variant and the maternally inherited exon 15 deletion were present in trans, establishing compound heterozygosity. When exome sequence data were reanalyzed, read depths derived from exon 15 were confirmed to be decreased to approximately 0.5 (Supplementary Fig. 1).
To assess the splicing consequence of c.1743-30_1932del, we performed RT-PCR using junction-spanning primers (Supplementary Table 2) on cDNA synthesized from total RNA of lymphoblastoid cells from the proband and her parents. RT-PCR revealed a predominant full-length product, but direct sequencing showed a minor transcript consistent with exon 15 skipping. To examine whether transcripts from the deletion allele undergo nonsense-mediated mRNA decay (NMD), we used the paternally inherited c.3430 C > G variant in exon 24 as an allele-specific marker and performed RT-PCR across exons 22–26. In the proband, sequencing of the RT-PCR product showed predominant expression of the G allele with only a faint residual C peak, whereas the father remained heterozygous and the mother lacked the variant (Fig. 1M). These results indicate that exon 15 skipping from the maternally inherited deletion allele leads to NMD, resulting in relative overrepresentation of the paternal allele.
c.1743-30_1932del was thus considered a loss-of-function variant, and was classified as likely pathogenic according to ACMG guidelines, meeting criteria PVS1 and PM2 [12]. In light of this, the missense variant c.3430 C > G was classified as likely pathogenic according to ACMG/AMP guidelines, meeting criteria PM1, PM2, PM3, and PP3 [12].
Comments (0)