
Remember me

You have full access to this article via your institution.
Addendum to: Nature Protocols https://doi.org/10.1038/s41596-024-00968-2, published online 15 March 2024.
The bioinformatics protocol by Wang et al.1 outlines the steps for efficiently identifying colinear blocks in intra- and inter-species BLASTP outputs using the Multiple Colinearity Scan Toolkit Version X (MCScanX)2. Part 2 of the protocol provides steps for downloading data directly from NCBI and preparing the necessary .gff and .blast files yielded from blast all-vs-all analyses. While using MCScanX, we (X.Z. and D.R.S.) discovered that Part 2 lacks an essential pre-processing step—the step required for determining whether there are multiple isoforms derived from alternative splicing. Indeed, when analyzing data downloaded directly from NCBI, it can be crucial to have a process known as transcript filtering to identify the longest transcript as the primary protein sequence. Similar issues could happen when researchers fail to use primary assembly (one haploid set) in diploid genome assemblies, because the alleles could be mistakenly treated as gene duplicates.
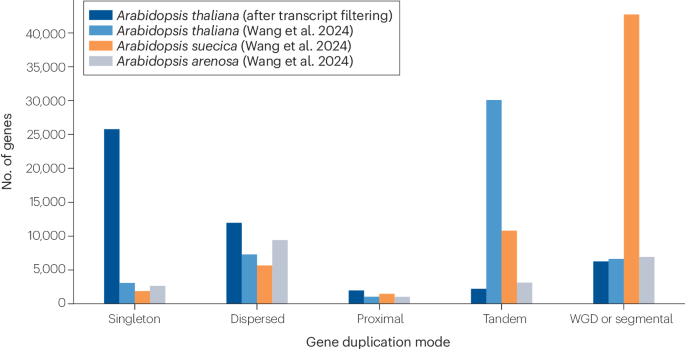
To avoid misprediction of gene duplicates, especially for genome data from NCBI or other online resources, it can be helpful to include a transcript filtering step3,4. This is particularly true when analyzing datasets of species with large numbers of duplicated genes. Without this step, the number of duplicate genes will be overrepresented. In Fig. 1, we show how using a transcript filtering step can dramatically impact the results of the analysis carried out in the protocol by Wang et al. Indeed, by following the MCScanX protocol and comparing Arabidopisis thaliana with and without transcript filtering, we found as many as 25,776 protein-coding genes categorized as singleton duplications after employing transcript filtering, compared to only 3,086 protein-coding genes when not filtering. Similarly, there are 11,948 protein-coding genes categorized as dispersed duplications when filtering vs. 7,297 protein-coding genes when not. Finally, there are only 2,216 genes categorized as tandem duplications after transcript filtering compared to 30,101 genes without filtering. This suggests that in the absence of transcript filtering, multiple isoforms from the same gene can be misinterpreted as tandem duplication events.
Fig. 1: Comparison of gene duplication modes among closely related Arabidopsis taxa, with and without transcript filtering. The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.This figure was adapted from Fig. 6 of the protocol1, where transcript filtering was not used (A. arenosa, A. suecica and A. thaliana without filtering are shown in grey, in orange and in light blue, respectively, as in Fig. 6 of the protocol1). A. thaliana after transcript filtering is shown in dark blue. Strikingly, it appears that tandem duplications are less prevalent in A. thaliana than A. arenosa, and singleton duplications represent an overwhelming proportion of gene duplications in A. thaliana compared to the other two species, when transcript filtering is incorporated into the workflow.
Figure 2 shows the visualization of the synonymous substitution rate (Ks) distributions of colinear genes in A. thaliana and Medicago truncatula before and after transcript filtering. We are not questioning the reliability of running the MCScanX algorithms but want to highlight potential issues when using the protocol, particularly the potential challenges when preparing input files in Part 2 (Steps 7–20). Overall, MCScanX is a useful tool for efficiently identifying colinear blocks and downstream evolutionary analysis, but additional work is needed for preparing the input data and running the tool.
Fig. 2: Visualization of the synonymous (Ks) substitution rate distributions of colinear genes in different genomes (A. thaliana and M. truncatula). The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Panels a and c are adapted from Fig. 5 of the protocol1. Red boxes highlight the differences in Ks before and after the transcript filtering. In Fig. 5 of the protocol, the authors used a different version of the A. thaliana genome (Araport11) as compared to the one demonstrated in the protocol (TAIR 10). Considering that the Araport11 version only has a few more protein-coding genes than the TAIR 10 version, we used the TAIR10 of A. thaliana to ensure data consistency throughout the protocol. Similarly, a high-quality, chromosome-scale reference genome for M. truncatula (MtrunA17r5.0-ANR) was used in this study, as the earlier Mt4.0v1 assembly was generated prior to the availability of long-read sequencing technologies.
We provide the following useful tips for increasing the utility of the protocol: as noted by the authors in Steps 11–13 and the Troubleshooting section, we found that when generating the correct .gff file, it is better to offer alternative options similar to the ‘mkGFF3.pl’ program in the MCScanX_protocol package. This is because the downloaded .gff can have different formats and it is important to convert it to the one MCScanX can read. We have found that the ‘gff2bed’ script from BEDOPS v2.4.41 (ref. 5), AGAT v1.6.1 (ref. 6) and the custom processing script on the ‘XX_feature_table.txt’ can help yield the .gff file for MCScanX. In terms of generating the .blast file at Steps 14–20, we found it is not efficient to prepare the ‘runBLASTP.sh’, especially when an all-against-all BLASTP is needed for each reciprocal genome pair. We have provided custom scripts with the MCScanX_Assistant tool at GitHub (https://github.com/zx0223winner/MCScanX_Assistant) to iterate the genome all-against-all BLASTP processing, which greatly improves the preparation step (see also the Supplementary Text S1–S4 in ref. 7).
These comments were well-received by the MCScanX team (Y.W., P.V.J. and A.H.P.) and a notice has been added to the external link of the protocol (http://bdx-consulting.com/mcscanx-protocol/) stating the following: “…the current stage lacks a transcript filtering step for handling multiple alternative splice isoforms per locus, which may lead to confusion among paralogous genes. To address this limitation, users are encouraged to utilize MCScanX_Assistant, which provides the necessary functionality”.
The MCScanX team further addresses the protocol’s lack of transcript filtering step as follows:
During the development of the original software, the MCScanX team recognized that alternative splicing could influence MCScanX results. To address this, the accompanying README file (https://github.com/wyp1125/MCScanX) explicitly states that “The xyz.bed file holds gene positions,” and the included example uses Arabidopsis thaliana gene symbols (e.g., AT1G01010) rather than transcript identifiers (e.g., AT1G01010.1). This guidance clearly indicates that users should supply gene-level names and coordinates—not transcript-level entries—in the .bed file. Furthermore, the original publication2 noted that “If a gene had more than one transcript, only the first transcript in the annotation was used.” Although the MCScanX toolkit did not include a dedicated transcript-filtering utility, the expected use of gene-level information in the .bed file was unambiguous.
In the MCScanX team’s subsequent research projects involving MCScanX, the .bed file was generated using custom scripts that performed transcript filtering, selecting either the first annotated transcript8 or the longest transcript9,10,11,12. The choice of filtering rule was made heuristically based on the specific biological question. To our knowledge, no consensus has yet emerged within the scientific community regarding an optimal transcript-filtering strategy, as this topic has not been comprehensively evaluated. Consequently, studies examining the effects of transcript filtering on gene colinearity and paralogous gene detection are both valuable and timely.
In the 2024 protocol1, the MCScanX team introduced a set of new automation and utility tools for MCScanX packaged separately as “MCScanX-protocol” (http://bdx-consulting.com/mcscanx-protocol/), including functions that enable direct processing of genome assembly files from NCBI. The current release of the mkGFF3.pl script within this package does not programmatically enforce transcript-filtering logic creating a potential pitfall for users unfamiliar with the original MCScanX documentation. The MCScanX team evaluated the consequences of this omission. Including all transcripts for a gene may lead to BLASTP matches among alternative isoforms of the same gene. Nonetheless, this has only limited impact on colinearity detection. As described in the original MCScanX publication2 the algorithm mitigates inflated local colinearity signals by collapsing consecutive BLASTP matches that share a common gene and whose paired genes are separated by fewer than five loci, retaining only the match with the smallest E-value. This behavior aligns with the results shown here in Fig. 1, where the numbers of WGD and segmental genes are nearly identical regardless of whether primary-transcript filtering is applied. Thus, the colinearity and synteny analyses presented in the protocol remain robust.
The more substantial effect of omitting transcript filtering is an overestimation of tandem gene pairs, caused by BLASTP hits among transcripts belonging to the same gene. This leads to inaccuracies in the gene-type counts reported in Step 26B and Fig. 6 of the protocol1; however, the MCScanX team did not detect tandem relationships among isoforms of the same gene in the corresponding phylogenetic analyses.
The MCScanX team encourages users of the protocol to consult both MCScanX_Assistant (https://github.com/zx0223winner/MCScanX_Assistant) and this Addendum for guidance on preparing an appropriate .bed file. The MCScanX team recognizes that transcript filtering by either the first annotated or the longest isoform remains an arbitrary choice and may introduce bias into downstream analyses. The field still lacks a benchmark or gold-standard dataset for objectively assessing the performance of gene-colinearity detection and gene-duplication mode classification. Developing such resources would be valuable for future research. The MCScanX team appreciated Dr. Zhang and Dr. Smith for their examination of the MCScanX-protocol package and for identifying areas for improvement. Both parties are actively collaborating to maintain and enhance both the MCScanX protocol and the MCScanX toolkit.
Comments (0)