Generation Scotland (GS)Overview
Generation Scotland is a Scottish family-based study with over 24,000 participants recruited between 2006 and 2011 [43]. Participants were aged between 18 and 99 years at recruitment, with a mean age of 47.5 years (SD 14.9). After exclusions (Supplementary Fig. 1), a total of 16,717 participants (9758 females and 6959 males) had measured blood-based DNAm (see Methods) and self-reported alcohol consumption data available (Supplementary Table 1 and Table 1). The mean units consumed in the week prior to completing the questionnaire and blood draw were 10.9 (SD 12.7, Supplementary Figs. 2 and 3). A total of 10,506 (62.8%) participants reported that this number was reflective of their usual drinking pattern with 1622 and 3756 noting it was less or more than they typically drink in a week (response unknown for N = 833).
DNA methylation
DNA methylation in blood at baseline (recruitment) was quantified for 18,413 Generation Scotland participants across three separate sets (NSet1 = 5087, NSet2 = 4450, NSet3 = 8876) using the Illumina MethylationEPIC (850 K) array. Individuals in Set 1 included a mixture of related and unrelated individuals. Set 2 comprised individuals unrelated to each other and to those in Set 1. Set 3 contained a mix of related individuals—both to each other and to those in Sets 1 and 2—and included all remaining samples available for analysis. Methylation data were processed across 121 experimental batches (NBatches. Set1 = 31, NBatches, Set2 = 30, NBatches, Set3 = 60).
Quality control details have been reported previously [44, 45]. Briefly, probes were removed based on (1) outliers from visual inspection of the log median intensity of the methylated versus unmethylated signal per array, (2) a bead count < 3 in more than 5% of samples, (3) ≥ 5% of samples having a detection p-value > 0.05, (4) if they pertained to the sex chromosomes, (5) if they overlapped with SNPs, and/or (6) if present in potential cross-hybridizing locations [46]. Samples were removed (1) if there was a mismatch between their predicted sex and recorded sex, (2) if ≥ 1% of CpGs had a detection p-value > 0.05, (3) if sample was not blood based, and/or (4) if participant responded “yes” to all self-reported diseases in questionnaires. A total of 752,722 CpGs remained after QC. Missing values were imputed using the mean of each CpG across all samples. Dasen normalization [47] was performed across all individuals.
Alcohol consumption data
Self-reported alcohol consumption was measured at baseline via questionnaires to obtain the number of units consumed in previous week (unit definition as per UK National Health Service: 8 g/10 ml of pure alcohol). Participants were also asked whether this was their usual drinking amount, or whether they had consumed more or less than normal. A total of 16,717 individuals had non-missing alcohol consumption data and methylation data—the rest of participants were excluded from this study (after exclusion, NSet1 = 4576, NSet2 = 4108, and NSet3 = 8033 individuals were left in sets 1, 2 and 3, respectively). Of these, 10,506 marked this quantity as representative of their typical weekly consumption, and 3756 stated this quantity was more than normal and 1622 less than normal (Supplementary Table 1, Supplementary Fig. 1).
Lothian Birth Cohorts of 1921 and 1936 (LBC1921 and LBC1936)Overview
LBC1921 and LBC1936 are longitudinal studies of ageing on individuals born in 1921 and 1936, respectively [20]. Study participants completed the Scottish Mental Surveys of 1932 and 1947 at approximately age 11 years old and were living in the Lothian area of Scotland at the time of recruitment in later life.
DNA methylation
Blood samples considered here were collected at around age 79 for LBC1921 and at around age 70 for LBC1936. DNA methylation was quantified using the Illumina HumanMethylation450K array, for a total of 692 (up to 3 repeated measurements from 469 individuals) and 2796 (up to 4 repeated measurements from 1043 individuals) samples from LBC1921 and LBC1936, respectively. Quality control details have been reported previously [48]. Briefly, probes were removed (1) if they presented a low (< 95%) detection rate with p-value < 0.01 and/or (2) if they presented inadequate hybridization, bisulphite conversion, nucleotide extension, or staining signal, as assessed by manual inspection. Samples were removed (1) if they presented a low call rate (< 450,000 probes detected at p-value < 0.01) and/or (2) if predicted sex did not match reported sex.
Self-reported alcohol consumption
Participants were asked about their usual alcohol consumption, including number of times alcohol is consumed per week, normal alcohol consumption, typical drink of choice, and glasses/pints consumed on average. From this information, alcohol consumption in units consumed per week was derived. A total of 436 and 895 individuals had non-missing alcohol consumption and methylome data available in LBC1921 and LBC1936 baseline, respectively, and were considered in this study.
ALSPACOverview
The Avon Longitudinal Study of Parents and Children (ALSPAC) is a cohort study of pregnant women resident in Avon, UK, with expected dates of delivery between 1 April 1991 and 31 December 1992 [22, 23]. Among these, 20,248 pregnancies were identified as being eligible and the initial number of pregnancies enrolled was 14,541 resulting in 14,062 live births and 13,988 children who were alive at 1 year of age. At the start of the study, mothers invited their partners to complete questionnaires. In total, 121,113 partners have provided data and 3807 are currently formally enrolled. As part of Accessible Resource for Integrated Epigenomic Studies (ARIES) [49, 50], a subsample ALSPAC children, mothers and partners had DNAm assayed using the Illumina Infinium HumanMethylation450 or MethylationEPIC Beadchip array from peripheral blood samples collected at multiple time points from birth to middle age. The present study used DNAm measured from peripheral blood samples collected from ALSPAC children at ages 15–17 (time point “15up”) and 24 (time point “F24”) [51], and from ALSPAC mothers and partners [52] 18 years after the study pregnancy. Study data were collected and managed using REDCap electronic data capture tools hosted at the University of Bristol. REDCap (Research Electronic Data Capture) is a secure, web-based software platform designed to support data capture for research studies [53]. Please note that the study website contains details of all the data that are available through a fully searchable data dictionary and variable search tool (http://www.bristol.ac.uk/alspac/researchers/our-data/).
DNA methylation
Illumina Infinium HumanMethylation450 and MethylationEPIC Beadchip arrays were used to assess genome-wide DNAm patterns in peripheral blood. Samples across different time points were distributed in a semi-random manner across slides in order to mitigate batch effects. Data pre-processing and normalization were performed using the R package meffil as previously described [50]. Samples with large numbers of undetected probe signals were removed, along with those that had sex or genotype mismatches. Probes undetected in more than 20% of samples were excluded.
Self-reported alcohol consumption
Alcohol consumption was measured as the estimated number of units consumed on average during the week the year before blood sample collection for DNAm analysis. Consumption was estimated multiplying alcohol intake frequency per week by intake quantity. Frequency was assessed by the question “How often do you have a drink containing alcohol”, with possible responses including “Never”, “Monthly or less”, “2–4 times a month”, “2–4 times a week”, and “4 or more times a week”. “Never” drinking respondents were considered non-drinkers and were included in all primary analyses. Quantity was assessed by asking the number of drinks consumed where “one drink referred to ½pint of beer/cider, a small (125 ml) glass of wine or a single (25 ml) measure of spirit”, each of which is roughly equivalent to one UK alcohol unit (8 g of ethanol).
Sister studyOverview
The Sister Study is a US-nationwide prospective cohort study of 50,884 women enrolled between 2003 and 2009; women were eligible for enrolment if they resided in the USA and were breast cancer-free themselves but had a biological sister who was previously diagnosed. As part of study enrolment when all women were breast cancer-free, women completed self-reported questionnaires and an in-home visit where a whole blood sample was collected. Information about obtaining data from the Sister Study can be found at: https://sisterstudy.niehs.nih.gov/English/coll-data.htm.
DNA methylation
Two case–cohort samples of women were selected for DNAm profiling. In 2014, blood DNA samples from 2878 self-identified non-Hispanic White women were assayed on the Infinium HumanMethylation450 BeadChip [54]. This sample included 1542 women who were diagnosed with breast cancer in the years following enrolment (mean time to diagnosis: 4 years). In 2019, blood DNA samples from 2599 self-identified Black (Hispanic and non-Hispanic) and non-Hispanic White women were assayed on the Infinium MethylationEPIC v1 BeadChip [55]. This sample included 999 women who were diagnosed with breast cancer in the years following enrolment (mean time to diagnosis: 5 years). Self-identified Hispanic and non-Hispanic Black women were over-sampled for DNAm profiling in order to maximize the racial and ethnic diversity of the MethylationEPIC sample.
For both DNAm samples, DNAm data were preprocessed using the ENmix software pipeline, which included background correction, dye bias correction, inter-array normalization, and probe-type bias correlation [56,57,58]. Samples were excluded if they did not meet quality control measure including bisulphate intensity < 4000, had greater than 5% of probes with low-quality methylation values (detection P > 0.000001, < 3 beads, or values outside 3 times the interquartile range), or were outliers for their methylation beta value distributions. In total, 178 participants from the HumanMethylation450 sample and 250 participants from the MethylationEPIC sample were excluded for not meeting quality control measures.
Alcohol consumption
Participants’ history of alcohol consumption was obtained within 1 year of blood draw as part of a baseline questionnaire for alcohol use. Women reported information including the age at which they started and stopped drinking alcohol. The frequency of alcohol consumption was reported as days per week, month, or year by decade of life. The alcohol use variable used in this study was a derived variable that represented the average number of drinks per week over the last twelve months.
EpiScore of alcohol consumption: who to train on, and how?
In an effort to assess the optimal cohort sample and feature space to train on, multiple EpiScores were assessed. The Generation Scotland cohort was divided into a training (sets 1 and 2, N = 8684) and a testing dataset (set 3, N = 8033). EpiScores were trained on the full training dataset, as well as just on the “normal week” drinkers (N = 5618). Further, EpiScores were trained on the full methylome (386,399 CpGs after limiting measured features to those also present in the Illumina 450 K array for wider applicability) or on a subset of 3999 epigenome-wide significant CpGs (P < 3.6 × 10−8) that have been previously linked to alcohol consumption in three separate studies not using Generation Scotland [9, 16, 25].
Elastic net penalized regression was used to train our EpiScores on log-transformed alcohol consumption + 1 (glmnet package in R, v4.1). CpG beta values in the training set were scaled to mean zero and unit variance ahead of elastic net, thus obtaining standardized regression effect sizes. The L1, L2 mixing parameter was set at α = 0.5, and tenfold cross-validation was performed to select the shrinkage parameter (λ) that minimized the mean cross-validated prediction error.
Predictive performance for each EpiScore was assessed by projecting the latter into the testing dataset by multiplying each CpG by its estimated weight and performing summation, scaling CpG beta values beforehand to mean zero and unit variance. Pearson correlation (r) of the EpiScore with measured log alcohol consumption + 1, as well as the incremental R2 upon the addition of the EpiScore to a linear regression model adjusting for age and sex, was then calculated. EpiScore statistical significance was assessed considering the marginal test for the beta in the linear regression model adjusting for age and sex (assessing whether beta is significantly different from zero).
Training the EpiScore in generation Scotland and testing in the Lothian Birth Cohorts, ALSPAC, and sister study
Having established that training on all individuals with self-reported alcohol consumption data (regardless of whether this pattern reflected a typical week or was more or less than normal), and on a pre-filtered set of CpGs, yields the better performing EpiScore, we next trained on the full Generation Scotland cohort (N = 16,717). As with the creation of previous EpiScores, elastic net penalized regression was used with α = 0.5 and tenfold CV. This EpiScore was then projected and tested on the Lothian Birth Cohorts of 1921 and 1936, ALSPAC, and the Sister Study. Its performance was again assessed via a Pearson correlation with self-reported alcohol consumption (log(x + 1) transformed) and the incremental R2 upon the addition of the EpiScore to a linear regression model adjusting for age and sex.
Sex-specific EpiScores
Sex-specific EpiScores were trained after matching sample sizes (thus ensuring larger sample sizes weren’t driving better prediction). Given that the smallest sex-stratified sample size was N = 6958 (males), we trained male-specific EpiScore on the full male sample set, a female-specific EpiScore trained on a random subsample of 6958 female participants, and a sex-agnostic EpiScore trained on equal numbers of males and females with overall sample size also being 6958 (NF = 3479, NM = 3479).
To assess performance, using the same metrics and testing LBC dataset described previously, we tested the resulting EpiScores in three different ways: (1) a sex-specific manner by which predictions are obtained using each testing sample’s sex-specific EpiScore, (2) an opposite-sex manner, by which the EpiScore trained on the opposite sex of the testing sample is used to obtain predictions, and (3) a sex-agnostic manner, by which all samples, regardless of sex, are predicted using the EpiScore trained on both males and females.
EpiScore and self-reported alcohol consumption associations in the Lothian Birth Cohorts
Associations between multiple phenotypes and self-reported alcohol consumption, as well as with our generated EpiScore trained on the full Generation Scotland cohort, were evaluated separately in LBC1921 and LBC1936. For each phenotype, linear regression models were run, adjusting for age, sex, and either self-reported alcohol consumption or the epigenetic predictor. Phenotypes considered included body mass index (BMI in kg/m2), hand grip strength (maximum of left and right hand measurements, in kg), self-reported years of education, self-reported smoking status (never smoker, ex-smoker, and current smoker), number of smoked packs per day, measured time taken to walk 6 m (in seconds), occupation-based social class (measured as social grades based on highest reached occupation [59]), and depression and anxiety scores (HADS-D and HADS-A total from the Hospital Anxiety and Depression questionnaire [60]). Associations with blood biomarkers cholesterol and triglycerides were also assessed.
Self-reported alcohol and alcohol EpiScore associations with self-reported prevalent disease were evaluated using logistic regression, adjusting for age and sex. These included CVD, stroke, neoplasm, high blood pressure, diabetes, and thyroid dysfunction. Associations with time to all-cause mortality were assessed using a Cox proportional hazards model with age and sex as covariates, using the survival R package (v3.5), with time to all-cause mortality or censoring as the survival outcome.
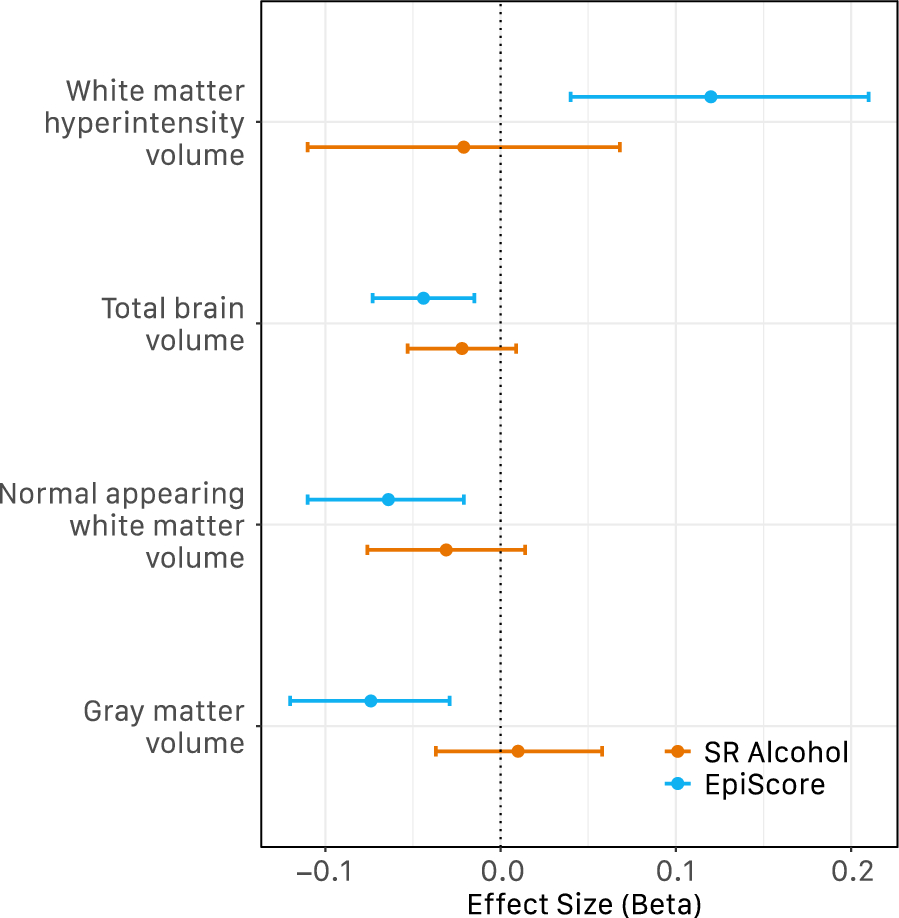
Finally, associations with multiple brain imaging phenotypes measured in LBC1936 were considered. Briefly, structural and diffusion tensor (DTI) MRI acquisition and processing in LBC1936 were performed at Wave 2 (age 73 years) according to an open-access protocol [61]. Total brain, grey matter and normal-appearing white matter (NAWM) volumes were calculated using a semi-automated multi-spectral fusion method [62]. Intracranial volume was determined semi-automatically using Analyze 11.0™. Total brain, grey matter, and white matter volume measurements were scaled to mean zero and unit variance, and associations with self-reported alcohol consumption and the alcohol EpiScore were assessed via linear regression, adjusting for age, sex, and intracranial volume.
Sample sizes varied for each phenotype considered given missing values arising from incomplete participant questionnaires (Supplementary Table 3). Association P-values were FDR corrected (using the Benjamini–Hochberg procedure) to account for multiple testing within each LBC cohort.
Variance components analysis and EWAS using BayesR+
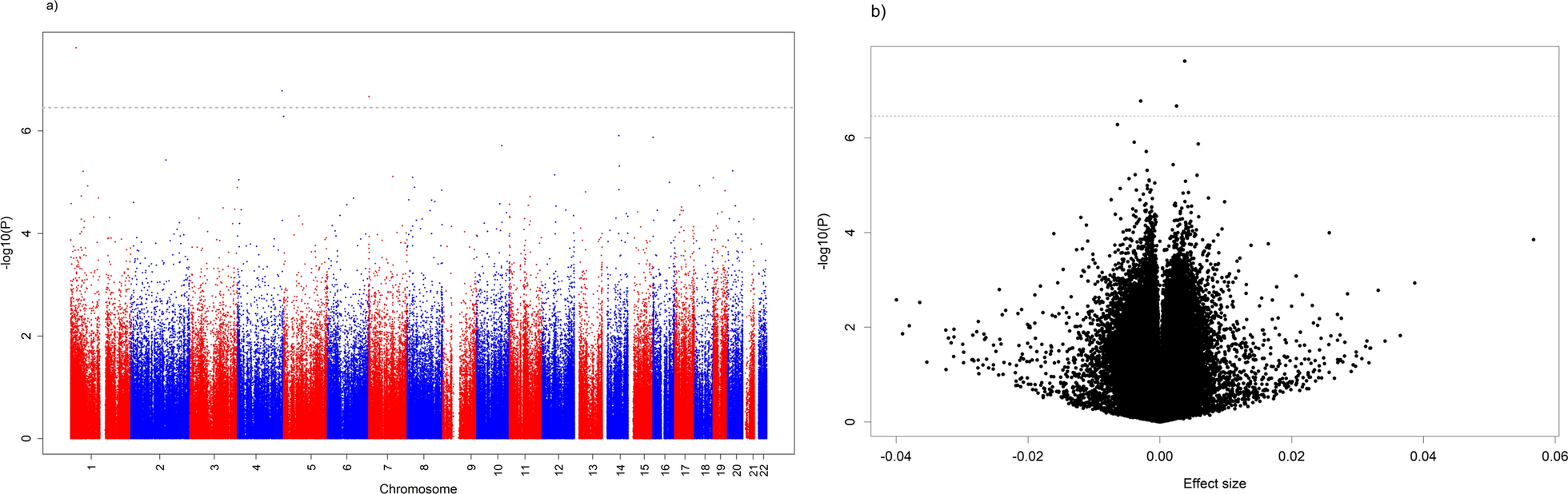
BayesR+ [26], a software implementation of a Bayesian regression modelling framework, which implicitly controls for white cell proportions, related participants, and other unknown confounders, was used to estimate the variance accounted for in alcohol consumption by methylation data, as well as estimate its association with each measured CpG (a total of 752,722). To remove the effects of age, sex, and smoking (via an EpiScore [7]), the input for BayesR+ was defined by the residuals of a linear regression model for alcohol consumption (log(x + 1) transformed) with those variables as covariates. CpG M-values were pre-corrected in a similar way, regressing out age, sex, smoking EpiScore, and batch. They were subsequently scaled to have mean zero and unit variance.
Full details of the BayesR+ modelling framework have been previously described [26]. Briefly, BayesR+ utilizes Gibbs sampling to generate draws from the posterior distribution conditional on the input data, setting prior mixture variances to 0.0001, 0.001 and 0.01, corresponding to possible small, medium, and large effect sizes of the CpGs considered (explaining 0.01%, 0.1% and 1% of the variance of the phenotype of interest, respectively). After a burn-in of 5000 draws, 10,000 draws were retained. Subsequently, a thinning of five draws was applied to reduce autocorrelation (i.e. 1000 iterations are used when reporting results for this analysis). The convergence of the hyperparameters was evaluated through the Geweke test [63], as well as assessing parameter values across iterations and assessing autocorrelation. For each probe, the proportion of iterations for which the probe was categorized as having a nonzero effect was calculated, this yielding the posterior inclusion probability (PIP). A PIP value over 0.95 (95%) was deemed to reflect an epigenome-wide significant CpG locus.
Variance components were estimated by the mean sum of squared standardized posterior effect sizes across the 1000 iterations. Individual effect sizes were estimated as the average across the 1000 iterations for each CpG. Models were run considering data for the full Generation Scotland cohort, as well as just the subset of “normal pattern” drinkers.


Comments (0)