
Remember me
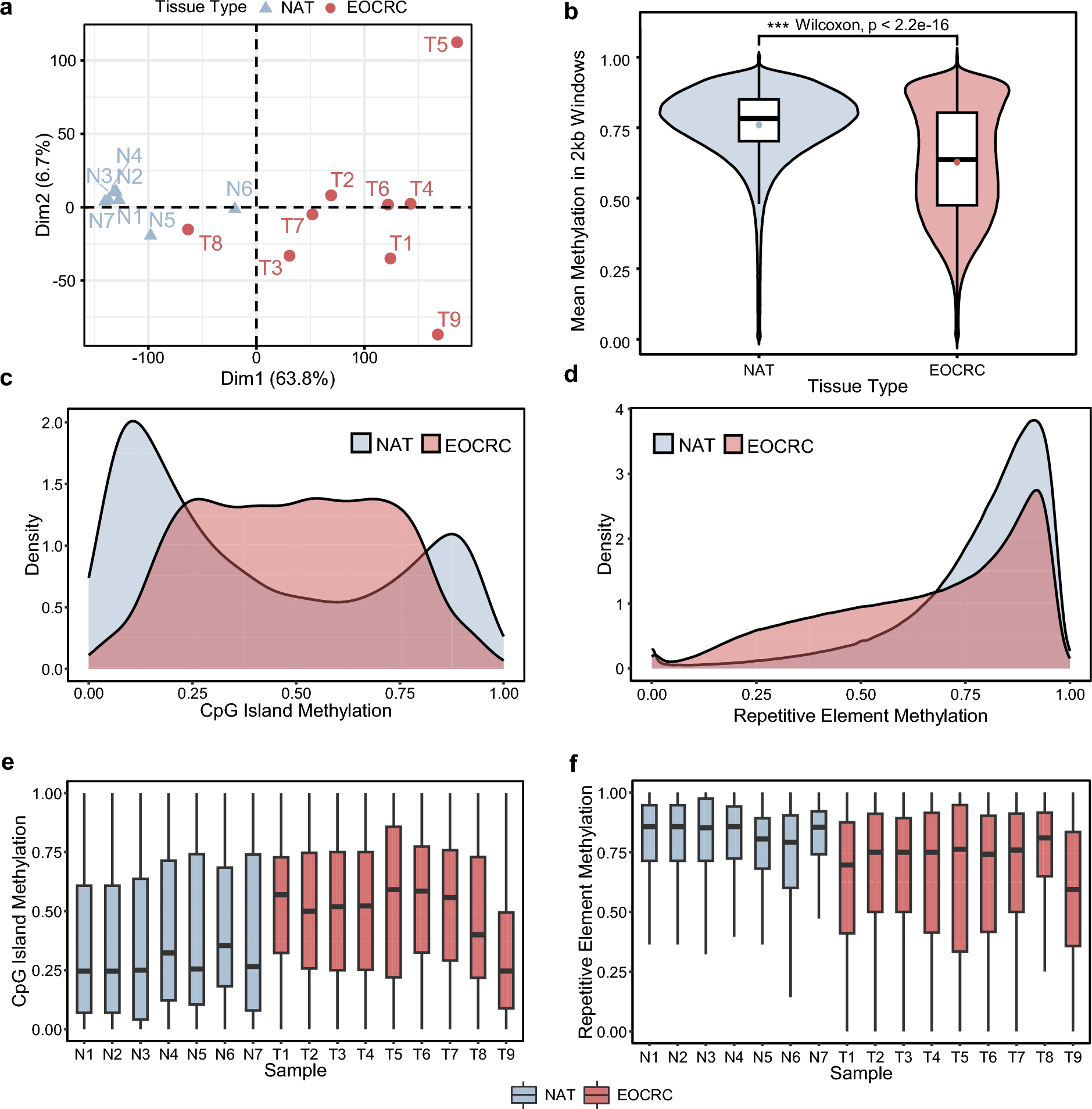

To first understand global methylation changes in EOCRC oncogenesis, we compared methylation between EOCRC tumors and matched NATs (Supplementary Table S1) across the entire genome by binning the genome into 2-kb sliding windows. EOCRC patients were selected using these inclusion criteria to exclude those arising from Lynch Syndrome or inflammatory bowel diseases (i.e., Crohn’s, ulcerative colitis): age of diagnosis < 50 years; mismatch repair (MMR)-proficient tumors; and nonhypermutated tumors (Supplementary Fig. S1). Based on the mean methylation of each sample at all 2-kb windows, we performed principal component analysis (PCA) and found that NAT clustered closely together, while EOCRC tumors were heterogeneous and distributed far away in PCA space from NAT, except for T8 (Fig. 1a). The clustering of sample T8 with NAT may reflect clinical confounders such as body mass index and comorbidities (e.g., type 2 diabetes), sample quality, and tumor heterogeneity. T8 was retained in subsequent analyses of the EOCRC cohort to more comprehensively reflect clinically relevant disease heterogeneity. Globally, tumor tissues were extensively hypomethylated (Fig. 1b), which is consistent with previous reports using a repetitive element LINE1 as a surrogate marker of global methylation in EOCRC [22,23,24]. In addition, the EOCRC tumors were highly hypermethylated at CpG islands compared to NAT (Fig. 1c), whereas repetitive elements (Alu, SINEs, LINEs) were overall hypomethylated (Fig. 1d). Similar trends were generally observed at a per-sample level, but with substantial heterogeneity in tumors (Fig. 1e and f).
Fig. 1
Global methylation characteristics of early-onset colorectal cancer. a Principal component analysis of methylation across 2-kb sliding windows for EOCRC tumor tissues (n = 9) and adjacent normal colonic mucosal tissues (n = 7). b Global methylation of aggregated samples across 2-kb sliding windows. EOCRC samples are globally hypomethylated, compared to NAT. c Density plot of CpG island methylation of aggregated samples showing extensive hypermethylation in EOCRC tissue. d Density plot of methylation at all repetitive elements in aggregated samples showing hypomethylation in EOCRC tissue. Boxplots of per-sample methylation at e CpG islands and f repetitive elements. CpG cytosine-guanine dinucleotide, EOCRC early-onset colorectal cancer, N1 to N7 normal adjacent tissue 1 to 7, NAT normal adjacent tissue, T1 to T9 tumor tissues 1 to 9
Differential methylation between EOCRC and normal adjacent tissueWe continued our unbiased comparison of EOCRC and NAT by performing differential methylation analysis using a circular binary segmentation algorithm from the tool Metilene [25]. To provide an overview of differential methylation, we performed both differentially methylated region (DMR) and differentially methylated CpG position (DMP) analysis in de novo mode. Regions differentially methylated between EOCRC and NAT groups were de novo annotated by circular binary segmentation and then filtered to keep only regions with ≥ 10 CpGs in the region and absolute methylation differences ≥ 0.1.
Filtered regions were evaluated using 2-dimensional Kolmogorov–Smirnov and Mann–Whitney U tests to yield a final DMR set with adjusted p value < 0.05. Most DMRs occurred in noncoding regions, including introns (47.9%) and intergenic regions (31.5%), but DMRs were also discovered in exons (12.1%) and promoters (6.8%) (Fig. 2a). Most noncoding region DMRs were hypomethylated, whereas promoters were more evenly split between hypomethylated and hypermethylated, favoring reduced methylation (Fig. 2c). These promoter methylation proportions resemble those previously observed in EOCRC using HM450K microarrays [26].
Fig. 2
Landscape of differential methylation in early-onset colorectal cancer. a Pie chart of differentially methylated regions (DMRs) according to gene location. b Pie chart of differentially methylated CpGs (DMPs) according to CpG island features. c Proportions of DMRs hypermethylated and hypomethylated according to gene location. d Proportions of DMPs hypermethylated and hypomethylated according to CpG island features. e Volcano plot showing significance and methylation difference for all promoters. Labels indicate gene names for the top 10 most significant hypermethylated and hypomethylated promoters. f Heatmap of the top 1% most variable hypermethylated and hypomethylated promoter DMRs. Tile color indicates row-wise normalized Z-scores. Columns are annotated by tissue type. CpG, cytosine-guanine dinucleotide; DM, differentially methylated; EOCRC, early-onset colorectal cancer; N1 to N7, normal adjacent tissue 1 to 7; NAT, normal adjacent tissue; N.S., not significant; T1 to T9, tumor tissues 1 to 9; UTR, untranslated region
To study DMPs, we used the Mann–Whitney U test with Bonferroni correction to evaluate significant differences between EOCRC and NAT groups for individual CpGs with an absolute methylation difference ≥ 0.1. DMPs occurred predominately in open seas (96.65%), while the remaining DMPs occurred in CpG shores (1.83%), shelves (1.45%), and islands (0.07%) (Fig. 2b). The majority of intergenic DMPs were hypomethylated (95.3%), which in combination with the high proportion of DMPs in intergenic regions (including repetitive elements) likely accounted for most of the global DNA hypomethylation found in EOCRC (Fig. 2d).
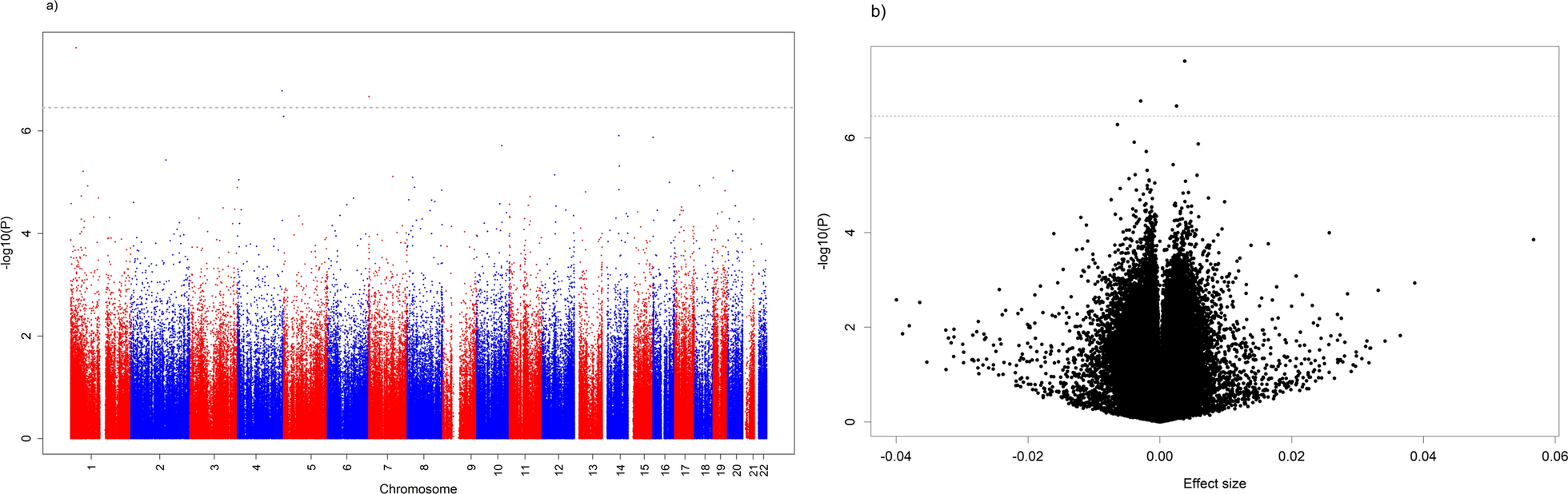
Despite the canonical view of transcriptional repression by DNA methylation, the relationship between methylation and gene expression is complex and position dependent. DNA hypermethylation correlates with reduced gene expression mainly at promoters. This correlation is tenuous or even reversed in other gene regions, obfuscating biological interpretation from methylome data alone [27]. Thus, we focused primarily on promoters in subsequent DMR analyses, performing another differential methylation study between EOCRC and NAT restricted to promoter regions. To ensure equal comparison between samples, we defined promoter regions as a window from 1000 bp upstream to 500 bp downstream of the transcription start site (TSS). In total, 1,073 statistically significant promoter DMRs (pDMRs) were identified, 694 (64.7%) of which were hypermethylated while 379 (35.3%) were hypomethylated in EOCRC, compared to NAT (Fig. 2e). Unsupervised hierarchical clustering of samples by the top 1% most variable pDMRs generally separated tumors from NAT, although the previous PCA outlier T8 clustered with the NAT group (Fig. 2f).
Methylation canyons overlap oncogenic pathway genes and are altered in colorectal cancerNext, because methylation canyons are linked to transcriptional dysregulation and oncogene expression in cancer, we performed de novo discovery of undermethylated regions (UMRs) using a previously developed Python script [28]. Adjacent UMRs within 500 bp were merged if the resulting region had a mean methylation value ≤ 0.1. We defined methylation canyons as UMRs with a methylation value < 0.1 and length ≥ 3.5 kb. To compare differences in methylation canyons between EOCRC and LOCRC, we included all currently available WGBS datasets of LOCRC tumors from TCGA (n = 5). Canyons were characterized by long valleys of low methylation, bordered by sharp plateaus of high methylation. As an example, one methylation canyon overlapped the promoter and body of the transforming growth factor beta receptor type 2 (TGFBR2) gene (Fig. 3a). Compared to NATs, we found that EOCRCs and, to a lesser degree, LOCRCs exhibited severe erosion of the downstream canyon border, suggesting increased transcriptional accessibility of TGFBR2.
Fig. 3
Methylation canyons overlap oncogenic pathway genes and are altered in colorectal cancer. Methylation canyons were defined as undermethylated regions with < 0.1 average methylation values and > 3.5 kb in length. a Smoothed line plot of methylation over the TGFBR2 canyon showing erosion of the downstream canyon border in EOCRC and LOCRC. Dashed lines represent approximate canyon borders. Gene transcript and CpG islands are indicated above; solid blue blocks represent the promoter and transcription start site, arrows represent the gene body and direction of transcription, and green blocks represent CpG islands. b Venn diagram of shared and unique gene canyon targets discovered in EOCRC and LOCRC. c KEGG pathway enrichment plot for selected top pathways comparing canyons discovered in EOCRC and LOCRC tissues. Undermethylated promoters (< 0.1 methylation) and promoter DMRs discovered in EOCRC are included as controls. d MSigDb oncogenic signatures enrichment plot for selected top pathways comparing canyons discovered in EOCRC and LOCRC tissues. e MSigDb cancer hallmarks enrichment plot for selected top pathways comparing canyons discovered in EOCRC and LOCRC tissues. CpG, cytosine-guanine dinucleotide; DMR, differentially methylated regions; EOCRC, early-onset colorectal cancer; LOCRC, late-onset colorectal cancer; N.S., not significant; Sig., significant; TGFBR2, transforming growth factor-β receptor type 2; UMP, undermethylated promoter
273 EOCRC-unique and 277 LOCRC-unique canyons were identified (Fig. 3b). To identify key pathways, we performed gene set overrepresentation analysis using three databases: KEGG (Fig. 3c), MSigDB Oncogenic Signatures (Fig. 3d), and MsigDB Cancer Hallmarks (Fig. 3e). As a control for non-canyon regions, we compared canyons to undermethylated promoters (UMPs, methylation < 0.1) unique to EOCRC (n = 1361) or LOCRC (n = 206) and to promoter DMRs detected in the EOCRC vs. NAT analysis (n = 1073). EOCRC-unique canyons exhibited strong enrichment in cancer-related pathways, notably the Wnt and TGF-β signaling pathways. Comparatively, LOCRC-unique canyons were significantly enriched only in TGF-β-related pathways. Although the biological and regulatory roles of these canyons require further exploration, these findings mirror previous reports that canyons are involved in oncogene regulation in solid tumors [17]. Interestingly, enrichment was mostly specific to EOCRC, despite approximately equal numbers of EOCRC-unique and LOCRC-unique canyons (Fig. 3b), suggesting that aberrant canyon methylation may be more associated with EOCRC rather than LOCRC tumorigenesis in these cohorts. Weak enrichment in the KEGG gene set occurred with EOCRC-unique UMPs, reflecting some overlap of promoters with canyons (Fig. 3c). However, promoter DMRs were not enriched in any of the cancer-related pathways enriched for in methylation canyons. Although case–control promoter DMR analysis is commonly used in DNA methylation cancer research, our findings show that DMR analysis alone is insufficient to capture gene set enrichment in biologically relevant pathways. This highlights the power of WGBS, which revealed biological insights undetected via traditional promoter DMR analysis.
Molecular basis of racial disparities between two cohorts of EOCRC patientsWe investigated differences between EOCRC patients of different racial/ethnic backgrounds by comparing two cohorts: Texas cohort, Hispanic (n = 8) or African American (n = 1); and TCGA cohort, Caucasian (n = 18). The TCGA cohort was selected based on the same inclusion criteria as the Texas cohort. To compare WGBS data (Texas cohort) to HM450K microarray data (TCGA cohort), we first reduced the whole-genome data by subsampling to only CpGs covered in the microarray. Although WGBS and HM450K data are well correlated in comparisons from the same sample [29], we addressed potential batch effects by performing quantile normalization. Contrast score distribution after normalization (measured by the false discovery rate-control tool Clipper [30]) was nearly symmetric around 0.0, suggesting removal of the batch effect (Supplementary Fig. S2). For both cohorts, we then assigned CpGs/probes to a gene promoter based on its location within the 1500-bp promoter window around the TSS. Normalized methylation values for each promoter were calculated by averaging all CpGs assigned to that promoter. pDMRs were defined as those with an absolute methylation difference ≥ 0.1 between cohorts. We then filtered for pDMRs with methylation values in tumors from racial and ethnic minority patients outside the 1st or 3rd quartiles of methylation values in Caucasian tissues (Methods) and used the nonparametric Wilcoxon ranked sum test to detect significant differences between cohorts (p < 0.05). We discovered 1168 remaining significant pDMRs between minority and Caucasian patients (Fig. 4a). Unsupervised hierarchical clustering of pDMRs showed race-specific patterns of DNA methylation, clustering cohorts separately from each other (Fig. 4b).
Fig. 4
Racial differences in promoter methylation between Caucasian and Hispanic/African American cohorts suggest a molecular basis for EOCRC disparity. Comparison between Hispanic and African American patients (Texas cohort) and Caucasian EOCRC patients (TCGA cohort). a Smoothed scatter density plot of 1,168 promoters differentially methylated in Hispanic and African American versus Caucasian EOCRC tissues. Color represents point density (blue, low; red, high). b Heatmap of significant differentially methylated promoters, normalized by row-wise z-score. Column annotations denote race. All patients had microsatellite-stable (mismatch repair-proficient) and nonhypermutated tumors. c Selected top enriched pathways relating to metabolism from all differentially methylated promoters. d Statistical analysis by interquartile range and Wilcoxon ranked sum test identifying genes with significant methylation differences between Hispanic/African American EOCRC tumors and Caucasian EOCRC tumors. Vertical lines indicate mean methylation values of each promoter in the Hispanic/African American (blue) or Caucasian (yellow) cohorts; distributions represent methylation values of all EOCRC tumors. e Same as d except for colorectal cancer-related genes. AA, African American; CRC, colorectal cancer; EOCRC, early-onset colorectal cancer; HDL, high-density lipoprotein; TCGA, The Cancer Genome Atlas
Because diet and metabolism are implicated in dysregulation of DNA methylation and are risk factors for CRC, we then performed gene set overrepresentation analysis to assess enrichment of race-associated pDMRs in metabolism-related pathways. Genes differentially methylated in minority EOCRC patients were significantly enriched in several metabolic pathways, including macroautophagy, a mechanism of metabolic homeostasis modulated by nutrient bioavailability (Fig. 4c). Macroautophagy is reprogrammed by tumor cells in response to increased metabolic demands and is also epigenetically dysregulated by diet [31, 32]. Enrichment in familial high-density lipoprotein deficiency (FHD) and type 1 diabetes pathways suggests a potential genetic component of aberrant methylation in metabolic pathways. Indeed, FHD is caused by apolipoprotein deficiency [33], and the apolipoprotein-encoding gene APOL3 was one of the top hypermethylated genes in minority, but not Caucasian, EOCRC patients (Fig. 4d). Other enriched pathways included amino acid and nucleotide metabolism gene sets, further supporting a nutrition-related epigenetic component in our minority cohort.
To further assess whether race/ethnicity-associated genes included canonical genes involved in CRC oncogenesis, we overlapped our pDMRs with the Human KEGG 2021 “Colorectal Cancer” pathway database and plotted the top CRC-related genes with racial/ethnic differences (Fig. 4e). Only four CRC-related genes (BIRC5, PIK3CD, RALGDS, and TGFB1) showed statistically significant methylation differences between racial groups, and the differences were substantially less than the differences noted for the previously identified top six genes (APOL3, MFAP2, etc.), except for TGFB1. This may suggest that in our racial/ethnic minority cohort, EOCRC disparities may be related more to auxiliary risk factors, such as metabolism, diet, and immune function, than to canonical CRC pathways. Together, our findings provide early insights for understanding the epigenetic basis for EOCRC racial disparities and characterize race/ethnicity-specific DNA methylation in our cohort.
Comments (0)