
Remember me
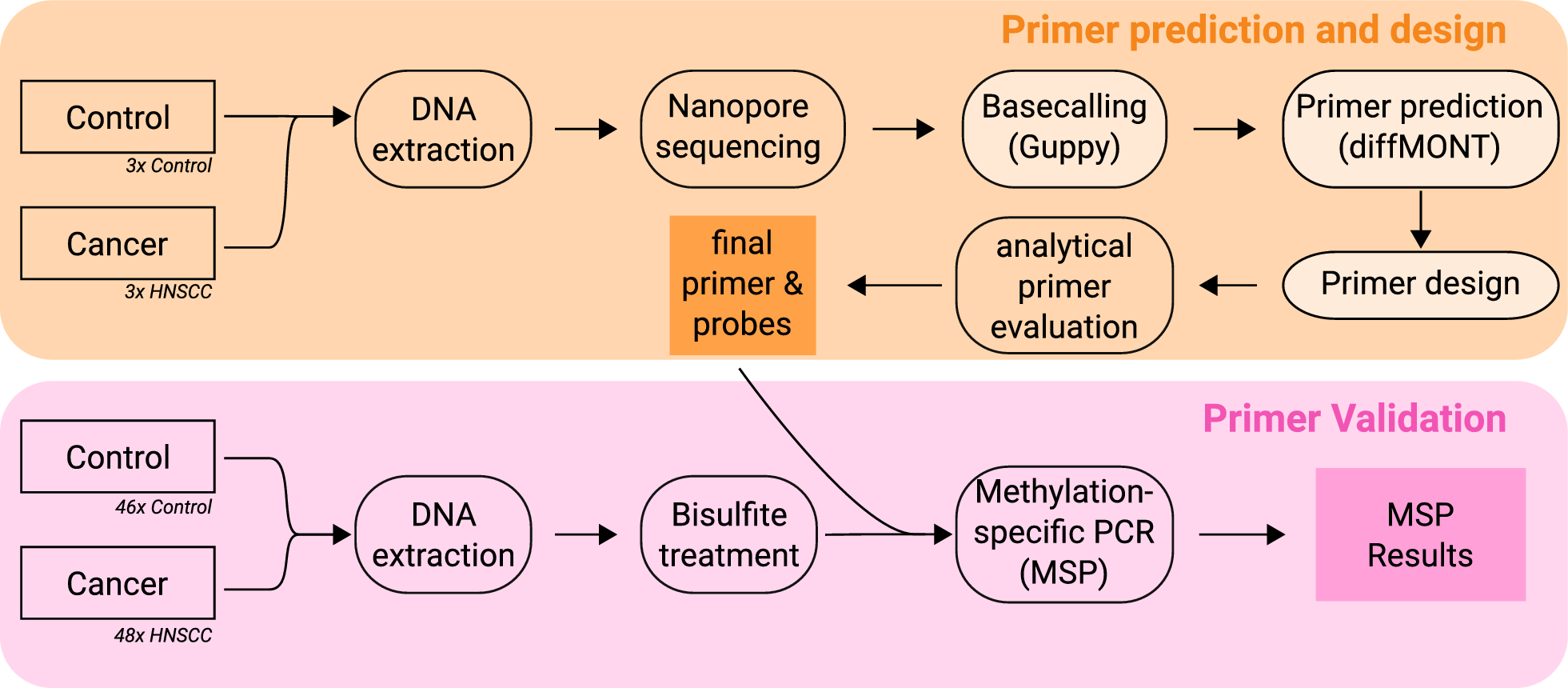

We developed a two-step, genome-wide workflow for the identification and validation of cancer-specific DNA methylation markers, as shown in Fig. 1. This approach is designed to be broadly applicable to a variety of cancer types and enables the systematic discovery of candidate regions with differential methylation patterns suitable for downstream MSP analysis.
Fig. 1
Based on nanopore sequencing data of three HNSCC and three control tissue samples, MSP primers were designed, and analytically evaluated. The best six primer pairs were chosen and validated with MSP assays for bisulfite-treated DNA extracted from 48 HNSCC and 46 control tissue samples. Steps, which were done purely in silico, are shown in a brighter orange (Basecalling, Primer prediction, Primer design)
Step 1: Marker discovery and primer design using nanopore sequencing This initial phase is relatively cost-intensive and involves sequencing a small number of cancer and control tissue samples using Oxford Nanopore Technologies (ONT) for the purpose of primer prediction and design. We recommend at least three cancer and three control samples, each sequenced on at least one ONT MinION flow cell. Genomic DNA is extracted from fresh-frozen tissue, and each sample is sequenced on at least one ONT flow cell. Methylation profiles are then analyzed using our in-house computational tool, diffMONT, which identifies differentially methylated regions (DMRs) across the genome and predicts suitable MSP primers for downstream primer validation. The overall cost for this discovery phase, including DNA extraction, library preparation, and sequencing, is currently estimated at approximately €7000.
Step 2: Validation by methylation-specific PCR The second phase of the workflow is designed for cost-effective validation of candidate markers in a larger patient cohort. This step requires standard molecular biology equipment and reagents. DNA is extracted from an extended set of cancer and control samples (we recommend at least 50 in total), followed by bisulfite conversion and methylation-specific PCR using the primers predicted in step one. The validated primers then allow for rapid screening of potential biomarkers at a low cost of less than €10 per sample and can be scaled depending on the availability of samples and the number of regions to be tested.
To demonstrate the feasibility and versatility of this approach, we applied the workflow to HNSCC samples. From the 100 available tissue samples, three HNSCC and three control samples were selected for nanopore sequencing and subsequent primer prediction. The remaining 94 samples, which included a diverse cohort in terms of sex, age, and tumor localization, were used for validation with MSP. The following sections detail each step of the workflow in the context of HNSCC, while the same principles may be transferred to other tumor types with minimal adaptation.
Primer prediction and design for HNSCC markersNanopore sequencing dataset results in 10 X sequencing depth on CpG islands with a tumor cell percentage of 70–80%
For nanopore sequencing and in silico marker prediction, three male HNSCC tissue samples with a high tumor cell percentage (T-0044-C, T-0085-C, T-0126-C) and three male control samples (T-0025-N, T-0045-N, T-0099-N) were chosen, as given in Table S1 and Fig. S1. For all tissue samples, genomic DNA could be extracted successfully. One of the six samples (T-0044-C) was tested positive for HPV 16.
To achieve a sequencing depth of 10 X on the CpG islands—which has been demonstrated as sufficient for reliable DNA methylation analysis using nanopore sequencing [38, 39]—adaptive sampling was applied to selectively enrich for sequencing depth on CpG islands. We focused on CpG islands, which account for \(\sim\)2% of the human genome [40], as they contain many of the regulatory methylation sites found in the genome [41]. Sequencing of the individual flow cells resulted in 4.11 Gb (T-0045-N) to 24.82 Gb (T-0025-N) output, which after basecalling resulted in a whole-genome coverage between 4.65 X and 8.44 X. Finally, for the CGI regions of each sample we gained a robust sequencing depth between 11.98 X (T-0126-C) and 28.66 X (T-0025-N), as given in Table 2.
In summary, we end up with a sufficiently reliable dataset for detecting differentially methylated regions with a tumor cell percentage above 70 % in the HNSCC samples, and a sequencing depth of at least 10 X on CpG islands.
Table 2 Characteristics and results of nanopore sequenced samples.In silico predicted regions are distributed over all chromosomes and overlap with both CpG islands and annotated genes
For the identification of potential epigenetic biomarker regions throughout the human genome, diffMONT was applied on the six nanopore sequenced samples to screen for hypermethylation in MSP-suitable regions in the three HNSCC tissue samples in comparison to the three control tissue samples.
Fig. 2
DiffMONT predicted a total of 50,479 regions. A The regions are distributed unevenly among the chromosomes. Data for 14,647 predicted regions located on unassembled chromosome patches are not shown. B 95.92% (48,420) of the predicted regions overlap with CpG islands. C 82.32% of the predicted regions overlap with annotated genes, 10.53% overlap not with genes but with promoters (500 nt upstream of transcription start site), and 7.15% with neither gene nor promoter. D Score distribution for the predicted regions. Higher scores result from high tumor sample methylation and coverage. 284 predicted regions have a score above 6000. E Following the diffMONT predictions, a manual selection process was conducted to determine the final primer choice
DiffMONT reported in total 50,479 regions distributed over all chromosomes, as shown in Fig. 2A. Of the predicted regions, 95.92% (48,420/50,479) overlap with annotated CpG islands in the genome, as shown in Fig. 2B. A high overlap with CpG islands was expected, because i) many regulatory methylation sites are located on CpG islands [41], and ii) sequencing was enriched for CpG islands. Of the 50,479 predicted regions, 82.32% (41,556/50,479) overlap with annotated genes, as shown in Fig. 2C. Each predicted region was scored by diffMONT based on methylation and coverage in the HNSCC samples, whereas a higher score indicates higher methylation and coverage. The score ranges from 203.98 to 9937.82 for the predicted regions, as shown in Fig. 2D. For the further analysis, only the top 284 regions with a score above 6000 were considered, as shown in Fig. 2E.
All high-scored predicted regions overlap with CpG islands but are in different genomic contexts—some overlap known HNSCC genes
Many of the individual MSP regions predicted by diffMONT are located very close to each other and have similar scores. This is expected, since we enriched for CpG islands, and cytosines throughout one CpG island are generally homogeneously methylated [42]. As a result, many of the predicted MSP regions overlap each other. To identify the most suitable primer sites for cancer detection, we selected only the highest-scoring MSP region from each overlapping cluster.
After this filtering step, 23 distinct MSP regions with a score > 6000 remained, which are distributed across 13 chromosomes, and all located on CpG islands, as given in Table S2.
CpG islands are often described in the context of gene promoters, however about half the CpG islands are not associated with annotated promoters [43]. Instead, they can be located upstream of genes, in exons, introns, downstream of genes, as well as intergenic. Similarly, our predicted MSP regions are distributed across the genome, found both in association with genes and in intergenic regions.
Fig. 3
The highest-scored predicted MSP region (204 nt, see also Table S2) is located in a CpG island on USP44
The highest-scored predicted region is located on a CpG island and overlaps both an exon and intron of the annotated gene USP44 (Ubiquitin carboxyl-terminal hydrolase 44), as shown in Fig. 3. This finding is supported by previously reports of the USP44 promoter described to be hypermethylated in nasopharyngeal carcinomas [44]. As expected, all reads from the control samples have a conserved low methylation in the predicted region and the reads from HNSCC samples are highly methylated.
Sample T-0085-C shows a slightly higher percentage of methylated reads compared to samples T-0044-C and T-0126-C, even though their total tumor cell content is similar. Interestingly, in the genomic context of the CpG islands the methylation pattern changes abruptly, and all control samples as well as the HNSCC samples show high methylation.
Another predicted region overlaps the bidirectional promoter of the genes ZNF529 and ZNF382, as shown in Fig. S2A. Methylation differences in bidirectional promoters have a double meaning in the context of gene expression regulation [45]. Foy et al. list promoter methylation of ZNF529 as potential marker candidate for OSCC [46].
In contrast, some of the predicted regions do not overlap with any annotated genes, as shown for the predicted region on chromosome 20, as shown in Fig. S2B.
Another 15 genes are overlapping with the the 23 highest-scored MSP regions, as given in Table S2. Among them, FLI1 was used by Gissi et al. for screening HNSCC based on DNA methylation in oral brushing samples [47].
Primer design based on in silico predicted regions
For the 23 distinct MSP regions, as given in Table S2, the goal was to design primers for at least one contiguous CpG island per chromosome, ensuring that the primer binding sites cover the target regions as precisely as possible and include at least three predicted CpG sites. For optimal design, however, it was necessary to adjust the primer boundaries by approximately ±5 nucleotides to optimize for the in silico calculated melting temperature (Tm). Of the 23 MSP regions, a subset was taken for further analysis, due to limited resources. The subset was selected by sorting the 23 regions by score, and taking the top 14 regions. Additionally, two regions on previously unrepresented chromosomes were added. As no primer design was possible for the selected region on chromosome 8, the next highest-scoring region on chromosome 8 was chosen. This selection resulted in 16 regions, for which primer candidates were selected for potential methylation-specific primer and probe design, resulting in 20 primer combinations, as given in Table S3. In the following, the designed primer pairs will be named based on the closest annotated gene, followed by PP (Primer Pair) and the score of the corresponding predicted MSP region (superscript), e.g., FLI1-PP1\(^\). If multiple primers were designed for a close related region, they were numbered consecutively (PP1, PP2,...).
Analytical evaluation of primer performance To determine the optimal annealing temperature for each primer pair a gradient PCR was performed as an initial step. Primer pairs were evaluated based on their amplification performance, characterized by a sigmoidal amplification curve, specific target amplification, low cycle threshold (C\(_T\)) values, minimal primer dimer formation, and melting curve profiles without unspecific products in negative controls (NC) and no-template controls (NTC).
Gradient PCR and confirmation of annealing temperature in primers
Out of 20 primer pairs tested, two primer pairs (FLI1-PP2\(^\) and PER1-PP1\(^\)) were excluded due to non-specific amplification. The primer pairs USP44-PP1\(^\) and LINC02579-PP1\(^\) were excluded due to non or inefficient amplification with high C\(_T\) values, respectively, indicating insufficient analytical sensitivity across the tested temperature range, as given in Table S3.
Amplification profiles allowed for the selection of optimal annealing temperatures for 16 primer pairs for further testing (RAI1-PP1\(^\), USP44-PP3\(^\), USP44-PP4\(^\), FAM43A-PP1\(^\), KREMEN2-PP1\(^\), BEND4-PP1\(^\), KCNB1-PP1\(^\), SMDP5-PP1\(^\), RAI1-PP2\(^\), EMX1-PP1\(^\), KCNB1-PP2\(^\), ZNF529-PP1\(^\), LHX8-PP1\(^\), URAD-PP1\(^\), FLI1-PP1\(^\), and USP44-PP2\(^\)).
The primer pair RAI1-PP2\(^\) was selected for further analyses, as both primer sets (RAI1-PP1\(^\) and RAI1-PP2\(^\)) showed comparable performance and targeted a similar predicted genomic region.
Following the gradient PCR, the previously determined annealing temperatures for all selected primer pairs were validated in a subsequent experiment. This involved amplification using a dilution series of the positive control (PC), including 10 ng and 1 ng input, alongside negative controls (NC) and no-template controls (NTC). We confirmed the analytical sensitivity of each primer pair and validated the annealing temperatures on the qPCR instrument by reproducing the C\(_T\) values observed during the gradient PCR, as given in Table S3.
Probe design and confirmation of probe functionality Additionally, for each of the validated amplicons, probes were designed. In total, 15 probes were designed for the most promising primer candidates (USP44-PP3\(^\), USP44-PP4\(^\), FAM43A-PP1\(^\), KREMEN2-PP1\(^\), BEND4-PP1\(^\), KCNB1-PP1\(^\), SMDP5-PP1\(^\), RAI1-PP2\(^\), EMX1-PP1\(^\), KCNB1-PP2\(^\), ZNF529-PP1\(^\), LHX8-PP1\(^\), URAD-PP1\(^\), FLI1-PP1\(^\), and USP44-PP2\(^\)). To evaluate probe performance, an initial gradient PCR (PC, NC, NTC as template) was again performed to determine the optimal annealing temperature for downstream applications and to assess probe functionality. Ideally, the C\(_T\) values obtained with the probe-based assays were expected to closely match those from the corresponding primer reactions with intercalating dye, indicating efficient probe hybridization and amplification without compromising assay sensitivity.
Of the 15 designed probes, an optimal annealing temperature could be directly established for 13 primer-probe combinations based on their amplification profiles. For USP44-PP3\(^\) and USP44-PP4\(^\), no probe signal was detected in the positive control when using the respective probes, as shown in Fig. S3. As a result, USP44-PP3\(^\) and USP44-PP4\(^\) were rejected.
Fig. 4
A Amplification curves of primer probe combinations show no probe binding in FAM43A-PP1\(^\) and KREMEN2-PP1\(^\) for 0.1 ng PC. BEND4-PP1\(^\) shows increased C\(_T\) values, especially for 0.1 ng PC. B All other primer probe combinations show specific amplification curves for all three dilutions. The curves illustrate high assay sensitivity, with consistent amplification of the target in the positive control, as well as high assay specificity, with no amplification in the negative controls. RFU—shows the resulting fluorescence signal in relative fluorescence units (RFU)
Building on this, the probe performance was evaluated by examining the amplification curves for a dilution series of the template (10 ng, 1 ng, 0.1 ng PC), as shown in Fig. 4. This step served to confirm both the annealing temperature settings and the analytical sensitivity of each assay. These assays demonstrated C\(_T\) values closely matching those obtained in the corresponding primer-only reactions, indicating that probe integration did not compromise assay performance and sensitivity, as given in Table S3.
Both FAM43A-PP1\(^\) and KREMEN2-PP1\(^\) show no amplification curve for 0.1 ng of input template. BEND4-PP1\(^\) showed increased C\(_T\) values especially for 0.1 ng of input template. Therewith, amplification is no longer reliably detectable. Consequently, these three primer-probe combinations were excluded from further analysis, as shown in Fig. 4A. The remaining ten primer probe combinations performed well in the dilution series PCR, as shown in Fig. 4B.
In the next step, a selection was made regarding which primer-probe combinations would be used for further clinical validation. Although ten assays demonstrated robust performance, the choice was guided by a cost-effective use of available tissue material. Six candidate markers were selected (KCNB1-PP2\(^\), ZNF529-PP1\(^\), LHX8-PP1\(^\), URAD-PP1\(^\), FLI1-PP1\(^\), and USP44-PP2\(^\)) based on their strong analytical performance and promising potential according to preliminary scoring results. The inclusion of additional markers remains a possibility for future analyses.
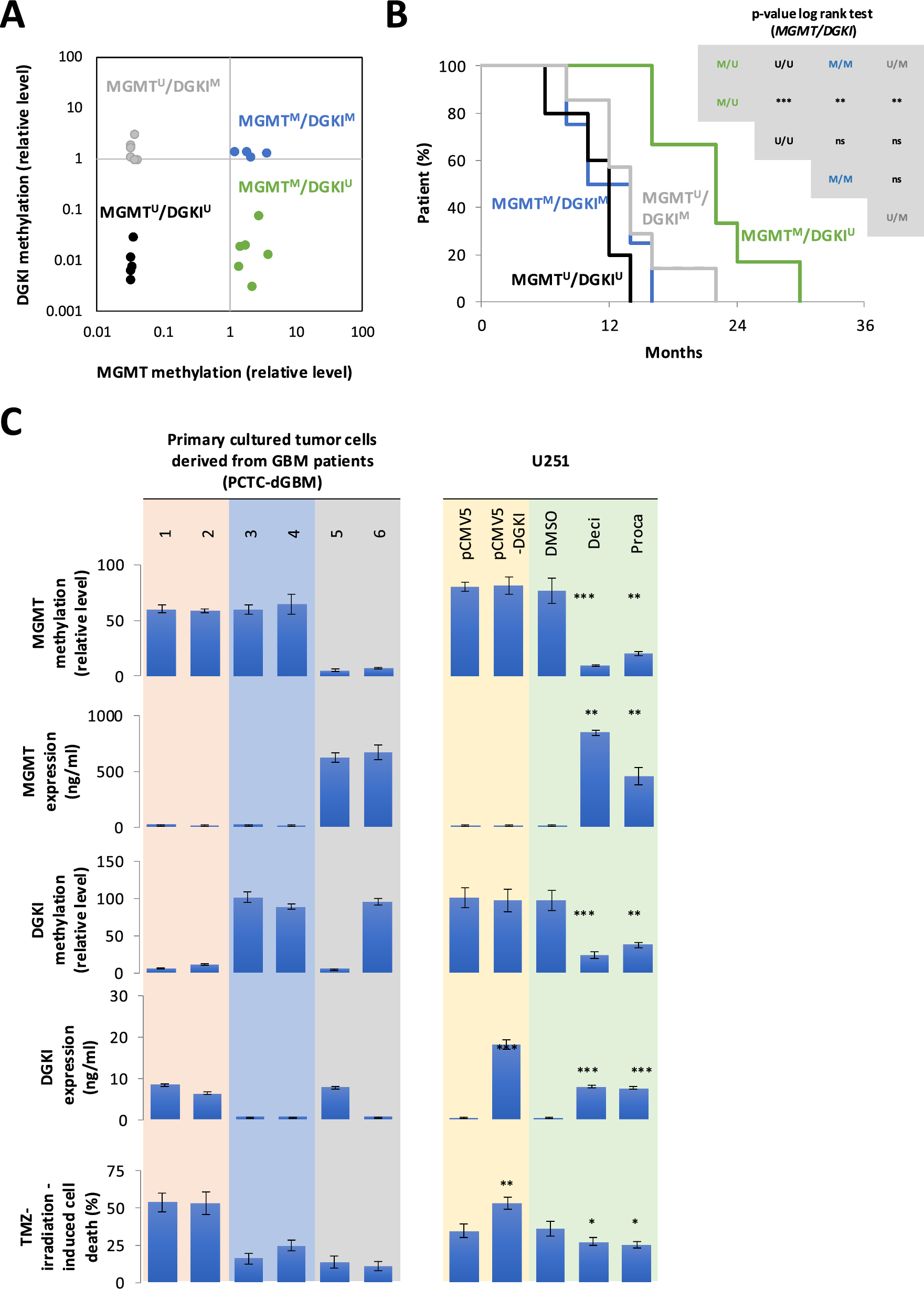
Validation of designed primers with MSPs demonstrates diagnostic potential in HNSCC and controlsFirst clinical validation of nanopore-derived primer pairs by MSP
After in silico prediction and analytical evaluation of the six primer pairs, their diagnostic utility was first tested in MSP assays for the three control and three HNSCC samples used for nanopore sequencing.
As expected, for each of the six regions, the MSP assay demonstrated higher \(\Delta\)C\(_T\) values for the control samples compared to the HNSCC samples, as given in Table 3. Despite a notable range of variability observed within both control and tumor samples (reaching up to 10.9 cycles for ZNF529-PP1\(^\) in control samples, and up to 19.62 for KCNB1-PP2\(^\) HNSCC samples), the minimum of \(\Delta\)C\(_T\) values from control samples was always higher than the maximum of HNSCC samples, with the notable exception of KCNB1-PP2\(^\) in patient T-0126-C.
Table 3 MSP \(\Delta\)C\(_T\) values for the predicted biomarker regions showed lower values in the HNSCC samples compared to the control samples for FLI1-PP1\(^\), LHX8-PP1\(^\), USP44-PP2\(^\), ZNF529-PP1\(^\), and URAD-PP1\(^\). The only exception was a high \(\Delta\)C\(_T\) for KCNB1-PP2\(^\) in sample T-0126-CTo compare our MSP primer \(\Delta\) C\(_T\) values with nanopore sequencing data and to assess individual patient-level methylation patterns, Fig. 5 illustrates the average 5mC methylation in the three HNSCC and three control samples, showing high methylation in HNSCC and low methylation in control samples across the final MSP primer regions—FLI1-PP1\(^\), LHX8-PP1\(^\), USP44-PP2\(^\), KCNB1-PP2\(^\), ZNF529-PP1\(^\), and URAD-PP1\(^\). The nanopore sequencing and MSP confirm the differential methylation for the regions detected by nanopore sequencing within these six samples. In case of primer pair KCNB1-PP2\(^\), the particular patient T-0126-C indeed has reduced methylation, comparable to control samples, as shown in Fig. 5D. Thus for these six samples, nanopore sequencing confirms the MSP results.
Fig. 5
Average 5mC methylation resulting from nanopore sequencing of three control (turquise, top) and three HNSCC (orange, bottom) samples shows high methylation in HNSCC and low methylation in control samples for the final MSP primer regions FLI1-PP1\(^\), LHX8-PP1\(^\), USP44-PP2\(^\), KCNB1-PP2\(^\), ZNF529-PP1\(^\), and URAD-PP1\(^\). The gDNA and biDNA sequences are shown for the sense strand in 5’\(\rightarrow\)3’ direction. The average methylation per cytosine is strand-specific
Selected primer pairs show diagnostic potential in clinical OSCC samples
Next, the validated primers were applied on additional clinical HNSCC tissue sample. As the nanopore sequenced samples used for primer design all originated from oral cavity and were therefore classified as oral squamous cell carcinoma (OSCC), a subset of HNSCC, a first validation was performed on 17 OSCC and 46 control tissue samples, excluding those which were used in nanopore sequencing.
We confirm the diagnostic potential for five of the six markers in additional OSCC samples, as shown in Fig. 6A. All primer pairs, except LHX8-PP1\(^\), show significant differences in the \(\Delta\)C\(_T\) values between OSCC and control tissue samples, confirmed by highly significant adjusted p values. The marker ZNF529-PP1\(^\) allows a nearly perfect classification into OSCC and control (Bonferroni corrected p value of 2.79\(\cdot\)10\(^\)), with a sensitivity, specificity, and AUC of 1, as shown in Fig. 6C.
The potential markers FLI1-PP1\(^\), USP44-PP2\(^\), KCNB1-PP2\(^\), and URAD-PP1\(^\) have a AUC above 0.8, and thus, all show good diagnostic accuracy. Generally, this type of analysis indicates a marker’s diagnostic potential (e.g., an AUC of 0.5754 for LHX8-PP1\(^\) suggests it may not be suitable as a diagnostic marker) [48]. While USP44-PP2\(^\) had the highest score in the in silico prediction and performed well in MSP of the sequenced samples, as given in Table 3, it was outperformed by ZNF529-PP1\(^\) and URAD-PP1\(^\) in the additional OSCC samples in terms of both Youden’s J and AUC.
Fig. 6
For each marker, an MSP assay was performed A on 17 OSCC and 46 control tissue samples, and B on 48 HNSCC (including the 17 OSCCs) and the same 46 control tissue samples. All markers except LHX8 show significant differences in the \(\Delta\)C\(_T\) values between control and OSCC samples as well as between control and HNSCC samples. Horizontal lines indicate optimal cut-points. Numbers on top are adjusted p values, three stars indicate a value below 0.01. C The marker ZNF529 outperforms the other markers with Youden’s J and AUC above 0.95 for both OSCC and HNSCC samples. Except for LHX8 in the HNSCC samples, all markers perform well with a Youden’s J above 0.5. Sensitivity (Sens.), Specificity (Spec.), and AUC were calculated for the optimal cutpoint (CP), which maximized Youden’s J statistic
Primers selected from OSCC show strong diagnostic performance across 48 clinical HNSCC samples
Although the primers were initially designed and tested using OSCC samples, a subset of HNSCC, they demonstrated robust diagnostic potential across a broader cohort of 48 clinical HNSCC tissue samples. This cross-applicability underscores the broader relevance and potential clinical utility of the selected methylation markers.
The transferability of the results from an OSCC subset of HNSCC samples to HNSCC tissue samples from different localizations was convincingly demonstrated in an MSP assay performed on 48 HNSCC samples (including the 17 OSCC samples analyzed in Fig. 6A) and the same 46 control samples as before, as shown in Fig. 6B. The results for the 48 HNSCC samples closely mirror those from the 17 OSCC samples, highlighting the robustness of the markers in an even more general context: For the primer pairs FLI1-PP1\(^\), USP44-PP2\(^\), KCNB1-PP2\(^\), ZNF529-PP1\(^\), and URAD-PP1\(^\) , the MSP results in significantly different \(\Delta\)C\(_T\) values for control and HNSCC tissues with adjusted p values below 0.01. Consistently with the OSCC samples, only the primer pair LHX8-PP1\(^\) does not show significant differences between the two conditions. Most prominent are the markers ZNF529-PP1\(^\) and URAD-PP1\(^\), which both have low \(\Delta\)C\(_T\) values with rather low variance (ZNF529-PP1\(^\): mean 4.94, standard deviation 2.55; URAD-PP1\(^\): mean 4.63, standard deviation 4.89) for the HNSCC samples. The marker FLI1-PP1\(^\) stands out by having high \(\Delta\)C\(_T\) values with low variance for the control samples (mean 18.42, standard deviation 2.31). These results are specifically striking, as the primers were predicted and designed based on three male-only HNSCC samples from oral cavity, while the samples used for Fig. 6B are highly diverse in sex, age, tumor localization and HPV status, as given in Table 1.
Thus, the markers FLI1-PP1\(^\), KCNB1-PP2\(^\), ZNF529-PP1\(^\), and URAD-PP1\(^\) show a high diagnostic performance with an AUC above 0.8, which classifies them as markers with very good diagnostic accuracy, as described by Šimundić [48]. Additionally, these four markers are characterized by a sensitivity above 0.7 combined with a specificity above 0.95, resulting in an Youden’s J statistic above 0.68, as shown in Fig. 6C.
In summary, the performance of an MSP assay with the described primers on 48 HNSCC and 46 control samples confirmed the generalizability of the predicted marker regions, as shown in Fig. 6B and C.
Comments (0)