
Remember me
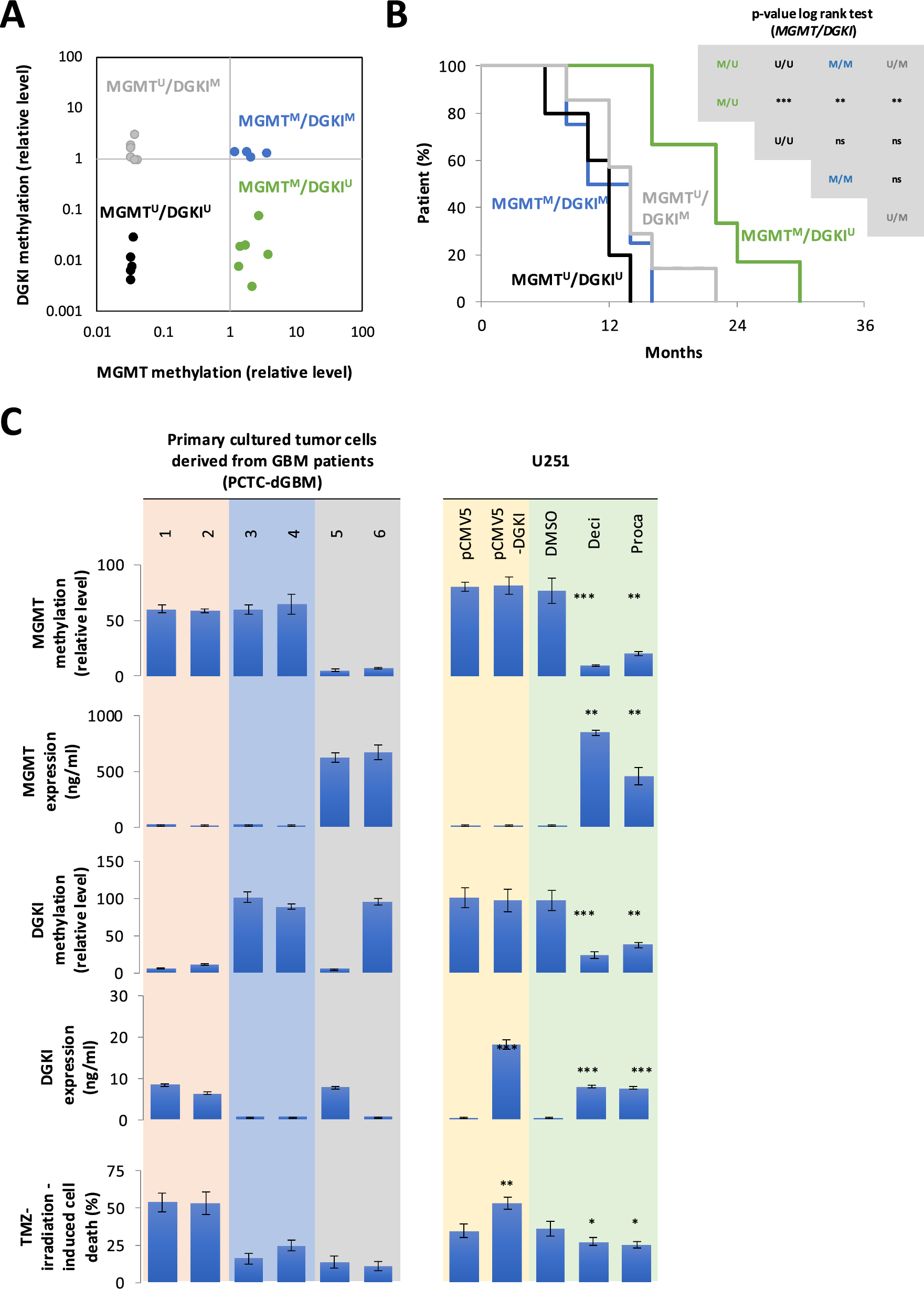
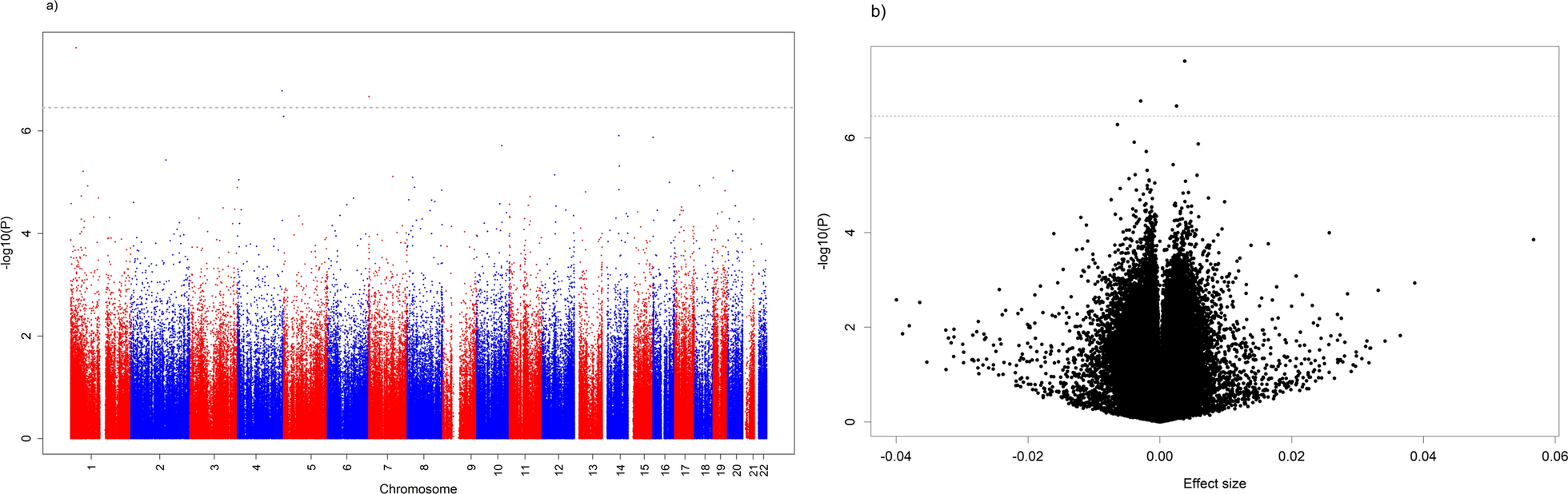

Previous studies demonstrated that DGKI promoter methylation defines a subset of MGMT-methylated GB patients with poor response to standard therapy, refining conventional MGMT-based stratification of GB patients [7, 8]. Based on these findings, a new short cohort of 22 GB patients was stratified into four molecular subgroups according to their methylated (M) or unmethylated (U) status: MGMTM/DGKIM, MGMTM/DGKIU, MGMTU/DGKIM, MGMTU/DGKIU (Table 1). Methylation levels of DGKI and MGMT were assessed using a quantitative methylation-sensitive restriction enzyme (qMSRE) assay (Fig. 1A). As expected, Kaplan–Meier survival analysis based solely on MGMT methylation confirmed that patients in the MGMT⁽M⁾ subgroup exhibited significantly improved overall survival (OS) compared to the MGMT⁽U⁾ subgroup (Supplementary Fig. 1 However, when stratification was refined using combined MGMT and DGKI methylation profiles, only the MGMT⁽M⁾/DGKI⁽U⁾ group showed a significant survival advantage over the three other subgroups (Fig. 1B). These findings validate DGKI promoter methylation as a poor prognosis marker in MGMT-methylated GB patients treated with temozolomide and irradiation.
Table 1 Characteristics of patients included in our two cohortsFig. 1
DGKI expression and methylation status are implicated in the response to standard glioblastoma therapy. A Twenty-two glioblastoma patients were stratified into four subgroups based on relative MGMT and DGKI methylation levels, as determined by the qMSRE assay. Each circle on the graph represents an individual patient. B Kaplan–Meier curves representing the overall survival of the four subgroups of GB patients obtained by considering the MGMT and DGKI methylation levels. C Graphs show the relationships between MGMT and DGKI methylation, their corresponding expression levels, and the percentage of cell death induced by TMZ and irradiation. Left panels correspond to six patient-derived glioblastoma primary cultures. Right panels correspond to U251 glioblastoma cells transfected with a DGKI-expressing plasmid (pCMV5-DGKI, Addgene, France), control plasmid (pCMV5) and treated (72 h) with two DNMT inhibitors: Decitabine (Deci, 1 M) and Procainamide (Proca, 0.5 M). T test was performed to estimate the significant difference between control and indicated conditions (p < 0.05*, p < 0.01** and p < 0.001***)
Given the known repressive effect of DNA methylation on gene expression, it was hypothesized that reduced DGKI expression in MGMTM/DGKIM GB cells may impair the efficacy of temozolomide- and irradiation-induced cell death.
To test this, TMZ-irradiation-induced cell death in 6 primary-cultured tumor cells (PCTC) was analyzed. The six PCTC derived from GB having different levels of DGKI/MGMT expression and methylation. As expected, PCTC-dGB with an MGMTM/DGKIU signature were sensitive to TMZ/IR-induced cell death (Fig. 1C, orange panel), whereas PCTC-dGB harboring the MGMTM/DGKIM signature (Fig. 1C, blue panel) exhibited resistant or limited sensitivity to TMZ/irradiation-induced cell death, like PCTC-dGB characterized by the unmethylated MGMT (Fig. 1C, gray panel).
To directly assess whether DGKI expression enhances therapy response, DGKI was re-expressed in the MGMT⁽M⁾/DGKI⁽M⁾ U251 GB cell line via plasmid transfection (pCMV5-DGKI, Addgene). U251 glioblastoma cell line was selected for this study due to its MGMTM/DGKIM methylation profile (Fig. 1C, yellow panel). DGKI overexpression in U251 cells transfected with the pCMV5-DGKI plasmid was validated by ELISA. (Fig. 1C, yellow panel). As shown in Fig. 1C, restored DGKI expression significantly increased TMZ/IR-induced cell death in U251 cells (Fig. 1C, yellow panel).
Together, these experiments indicate that DGKI expression contributes to increased sensitivity to TMZ and irradiation-induced cell death.
Currently used DNA demethylating agents are inefficient at demethylating DGKI without demethylating MGMTGiven that DGKI expression enhances TMZ- and irradiation-induced cell death in MGMTM/DGKIM cells, it was hypothesized that selective demethylation of DGKI could improve the therapeutic response in this molecular subtype. To test this, effects of two non-selective DNA demethylating agents were assessed on MGMT and DGKI methylation levels, as well as on the extent of TMZ- and irradiation-induced cell death. Thus, two distinct DNMT inhibitors were used: the nucleoside analog Decitabine [11], which incorporates into DNA and traps all DNMTs, and the catalytic inhibitor procainamide [12], which specifically blocks the active site of DNMT1.
First, qMSRE assays confirmed that Decitabine and Procainamide induced the demethylation of both DGKI and MGMT genes (Fig. 1C, green panel), consistent with their non-selective mechanism of action. In this context, MGMT and DGKI expression increased but TMZ/IR-induced cell death decreased (Fig. 1C, green panel). In other words, despite promoting DGKI demethylation and re-expression, these agents failed to enhance TMZ sensitivity, likely due to concomitant reactivation of MGMT, a key mediator of TMZ resistance.
Design of EXDDNMT1/ELK1, a peptide that selectively demethylates DGKI without affecting MGMTSince both non-selective DNA demethylating agents failed to specifically target DGKI without affecting MGMT, efforts were directed toward identifying a molecule capable of selectively demethylating the DGKI promoter. Based on previous findings from our group and others, DGKI methylation was hypothesized to be regulated through a DNMT1–transcription factor interaction [13,14,15,16,17,18,19], specifically by a mechanism called TF-directed DNA methylation. To identify TFs potentially interacting with DNMT1 to regulate DGKI methylation, the PROMO predictive software (https://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3) was used. Thus, 22 TFs were identified (Supplementary Fig. 2). Among these 22 TF, only two have been reported to interact with DNMT1, based on the work of Hervouet et al. (2010): cEBPα and ELK1 [15] (Fig. 2A).
Fig. 2
Design of a DNMT1/ELK1 interaction inhibitor to demethylate DGKI without demethylating MGMT. A Identification of DNMT1/ELK1 as key regulators of DGKI methylation. The schematic “metro map” illustrates the stepwise strategy used to predict candidate regulators of DGKI methylation. The graph shows the effect of siRNA-mediated knockdown of selected factors on DGKI methylation levels. T test was performed to estimate the significant difference between control and indicated conditions (p < 0.05*, p < 0.01** and p < 0.001***). B The computational model depicts the interactions between a selected region of DNMT1 (349–500) shown in blue and in green, and the structured region 1–95 of ELK1 displayed in orange. The green region represents the 461–471 amino acids regions of DNMT1 interacting with ELK1. The model confidence was calculated using a combination of pTM and ipTM score, as reported in Materials and Methods. C The ChIP and ReChIP experiments were performed 24 h after the cells were treated with 0.5 µM of each indicated peptide. Graph illustrates the impact of EXDDNMT/ELK1 and EXDDUP on DNMT1 and ELK1 enrichment on DGKI promoter. IgG was used as negative control on ChIP and reChIP experiments. T test was performed to estimate the significant difference between control and indicated conditions (p < 0.05*, p < 0.01** and p < 0.001***). D qMSRE were performed 24 h after the cells were treated with 0.5 µM of each indicated peptide. Graph illustrates the impact of EXDDNMT/ELK1 and EXDDUP on the DGKI and MGMT methylation levels. T test was performed to estimate the significant difference between control and indicated conditions (p < 0.05*, p < 0.01** and p < 0.001***). E ELISA was performed 24 h after the cells were treated with 0.5 µM of each indicated peptide. Graph illustrates the impact of EXDDNMT/ELK1 and EXDDUP on DGKI expression T test was performed to estimate the significant difference between control and indicated conditions (p < 0.05*, p < 0.01** and p < 0.001***)
To assess whether DNMT1, CEBPα, or ELK1 contribute to DGKI methylation, DGKI methylation level was analyzed following siRNA-mediated silencing of each factor (Supplementary Fig. 3). qMSRE analysis revealed that silencing of DNMT1 or ELK1 reduced DGKI methylation levels, whereas CEBPα knockdown had no detectable effect (Fig. 2A). These observations support the hypothesis that DGKI methylation is regulated through functional interaction of DNMT1 and ELK1.
AlphaFold2, the deep learning-based structural prediction tool developed by DeepMind, was employed to identify and design a peptide targeting the DNMT1/ELK1 interaction. Initially, protein regions were analyzed and selected to enable accurate prediction of the DNMT1/ELK1 interaction interface. For DNMT1, residues 349–1130 were selected for complex prediction, corresponding to its structured core excluding the catalytic C-terminal domain (residues 1130–1600). ELK1 remains poorly characterized structurally; however, Mo et al. identified its DNA recognition domain (residues 1–94) as critical for DNA interaction [20]. These considerations led to the hypothesis that DNA recognition domain of ELK1 mediates its interaction with DNMT1. Accordingly, this domain was selected for AlphaFold-based structural prediction. An initial AlphaFold prediction using DNMT1 (349–1130) and ELK1 (1–95), although low in confidence (~ 30%) and not shown here, suggested a potential ELK1 interaction site within residues 350–500 of DNMT1. A second prediction was therefore performed, focusing on this region. In the second model, a very high confidence score (82%) was observed, suggesting high reliability in predicting ELK1 interaction with DNMT1 residues 461–471 (Fig. 2B). In other terms, these data suggest that the 461–471 amino acid region of DNMT1 is engaged in the DNMT1/ELK1 interaction. Moreover, the predicted interaction suggested that electrostatic forces and hydrogen bonding may occur between DNMT1 residues N462 and E463 and ELK1 residues D58 and K75. (Supplementary Fig. 4). Additionally, this prediction also indicated that W454 residues of DNMT1 could interact with phenylalanine F81 of ELK1 through π-stacking interactions (Supplementary Fig. 4). Based on these findings, a peptide mimicking DNMT1 residues 461–471 was synthesized. To facilitate intracellular delivery and nuclear localization, a cell-penetrating sequence and a nuclear-targeting signal were appended. This peptide construct was subsequently named EXDDNMT1/ELK1.
Then, ChIP and reChIP experiments showed that EXDDNMT1/ELK1 inhibited both DNMT1 and ELK1 recruitment to DGKI gene, compared with a control peptide (EXDctrl) (Fig. 2D).
In comparison, a peptide targeting DNMT1/PCNA/UHRF1 (EXDDUP), essential for DNMT1 activity and known for his unspecificity, did not alter of DNMT1 and ELK1 recruitment to DGKI gene (Fig. 2D), showing the specific effect of EXDDNMT1/ELK1. In addition, qMSRE assays showed that EXDDNMT1/ELK1 decreased DGKI methylation level without affecting MGMT methylation level (Fig. 2E).
In conclusion, EXDDNMT1/ELK1 inhibits both DNMT and ELK1 recruitment to DGKI gene, resulting in reduced DGKI methylation without affecting MGMT methylation level.
EXDDNMT1/ELK1 increases anti-GB effect of TMZ in PDX model of GB without promoting pro-tumoral phenotypesEXDDNMT1/ELK1 was next evaluated for its impact on TMZ/irradiation-induced cell death, doubling time, invasion and global level of DNA methylation, three cancer hallmarks and one signature associated with GB tumorigenicity [21,22,23], were carried out.
Treatment with EXDDNMT1/ELK1 significantly increased TMZ/IR-induced cell death, while having no detectable effect on invasion index, doubling time and global DNA methylation level of U251 (Fig. 3A). On the contrary, Decitabine, Procainamide and EXDDUP, various non-specific methylation inhibitors reduced TMZ/IR-induced cell death. This effect could be explained by multiple molecular mechanisms triggered upon DNMT inhibition, notably the re-expression of MGMT, a DNA repair enzyme that counteracts DNA damage. To quantify the therapeutic index of each compound, we developed a Score of Modulation of Cancer Hallmarks (SMoCH). Notably, EXDDNMT1/ELK1 was the only one DNA demethylating agent presenting a positive SMoCH (Fig. 3A), indicating that it suppressed more cancer hallmarks than it promoted, unlike other tested DNA demethylating agents. This supports its relevance as a candidate for personalized therapy targeting GB with the MGMTM/DGKIM signature.
Fig. 3
EXDDNMT1/ELK1 increases the TMZ/IR-induced cell death and unchanged the cellular proliferation, invasion and migration. A Graphs show the effects of various DNA hypomethylating agents on TMZ/irradiation-induced cell death, cell doubling time, migration index, global DNA methylation levels, and the modulation score of cancer hallmarks. (SMoCH) (− 1 when the peptide/treatment enhanced a cancer hallmark, 0 when peptide/treatment did not modify a cancer hallmark and + 1 when the peptide/treatment inhibited a cancer hallmark). Experiments were performed 72 h after the cells were treated with 1 µM of Decitabine (Deci), 0.5 µM of Procainamide (proca) of 0.5 µM of each indicated peptide. T test was performed to estimate the significant difference between control and indicated conditions (p < 0.05*, p < 0.01** and p < 0.001***). B Schematic overview of the treatment protocol in mice. Upon reaching a tumor volume of ~ 100 mm3, mice were randomly assigned to the indicated treatment groups. TMZ (25 mg/kg) was administered intraperitoneally (i.p.), and EXDDNMT1/ELK1 was delivered intratumorally (i.t.). The photograph shows resected tumors after 3 weeks of treatment. The graph depicts tumor volume responses across three distinct patient-derived xenograft (PDX) models. T test was performed to estimate the significant difference between control and indicated conditions (p < 0.05*, p < 0.01** and p < 0.001***). C Graph shows changes in MGMT and DGKI methylation levels following treatment. T test was performed to estimate the significant difference between control and indicated conditions (p < 0.05*, p < 0.01** and p < 0.001***)
The anti-tumor efficacy of EXDDNMT1/ELK1 was further validated in TMZ-treated PDX model of GB. Subcutaneous implantation of three MGMT⁽M⁾/DGKI⁽M⁾-classified PCTC-dGB lines was performed in nude mice. Once tumor volume reached 100 mm3, mice were selected to receive in situ injection of control solution, TMZ, EXDDNMT1/ELK1, or TMZ + EXDDNMT1/ELK1 (Fig. 3B). Results showed that only TMZ + EXDDNMT1/ELK1 treatment led to a reduction in tumor volume (Fig. 3B). In addition, tumors treated with EXDDNMT1/ELK1 were characterized by a decrease in DGKI methylation, while MGMT methylation level was unchanged (Fig. 3C).
Collectively, these data indicated that EXDDNMT1/ELK1 enhances the TMZ anti-tumor effect by demethylating DGKI in GB cells harboring the MGMTM/DGKIM signature.
EXDDNMT1/ELK1 prevents the TMZ resistance acquisitionThe potential impact of prolonged TMZ exposure on DGKI methylation was next examined. To address this at cellular level, three GB cell lines were exposed to TMZ at their respective IC₅₀ concentrations for eight weeks. All cell lines acquired TMZ resistant and only A172-TMZ8w cells displayed an increase in DGKI methylation level (Fig. 4A). Interestingly, MGMT methylation level was unchanged under these conditions in three cell lines (Fig. 4A). To investigate whether TMZ treatment might modulate DGKI methylation level in patients, two series of experiments were performed. DGKI methylation levels were first assessed in a cohort of 15 GB patients who underwent two surgical resections: one before treatment (R1) and one at recurrence after TMZ therapy (R2) (Table 1). Importantly, DGKI methylation level increased in six of the fifteen patients between the two surgical resections, remained unchanged in eight of the fifteen patients and decreased in only one patient (Fig. 4B). In addition, MGMT methylation levels remained unchanged between first and second resections in these patients. (Fig. 4B). Then, based on these results, methylation profile evolution between the two resections was examined. For this, genes with methylation levels below 50% were classified as unmethylated, while those with levels equal to or above 50% were classified as methylated. Thus, five patients have a methylation profile that evolved from MGMTM/DGKIU to MGMTM/DGKIM between the two resections (Fig. 4C). This means that five patients shifted from a favorable to an unfavorable prognosis for response to standard anti-GB therapy between the two tumor resections (Fig. 4C). Conversely, one patient shifted from an unfavorable prognosis (MGMTM/DGKIM) to a favorable prognosis (MGMTM/DGKIU) for response to standard anti-GB therapy between the two tumor resections (Fig. 4C).
Fig. 4
EXDDNMT1/ELK1 limits the TMZ resistance acquisition. A Graphs depict MGMT (blue) and DGKI (orange) methylation levels in glioblastoma cell lines (U373, U87, and A172) before and after 8 weeks of temozolomide treatment. Sensitivity and resistance was defined based on TMZ IC₅₀ values (gray). T test was performed to estimate the significant difference between control and indicated conditions (p < 0.05*, p < 0.01 ** and p < 0.001***). B Graphs illustrate the MGMT (blue) and DGKI (orange) relative methylation levels in tumor reSect. "Introduction" (R1) and 2 (R2) derived from 15 GB patients. C Evolution of DGKI and MGMT methylation levels between the first (R1) and second (R2) tumor resections in the 15 glioblastoma patients included in the study. Black circle represents a methylated gene and open circle represents an unmethylated gene. Green panels underline the presence of the MGMTmethyalted/DGKIunmethyalted favorable signature. D Graphs illustrate the evolution of the MGMT and DGKI methylation levels in cfcDNA of five GB patients. E IC50 curves of temozolomide for A172 cells (black line), A172 cells treated during 8 weeks with TMZ (A172-8w, gray line) and A172 cells treated during 8 weeks with TMZ and EXDDNMT1/ELK1 (A172-8w + EXDDNMT1/ELK1, dark red line)
In a second investigation based on 5 new GB patients, evolution of DGKI and MGMT methylation levels in cell-free circulating DNA (cfcDNA) during treatment in five GB patients was analyzed. Analysis of DGKI and MGMT methylation in cfcDNA is supported by the well-established idea that cfcDNA partially reflects tumor-derived DNA (tDNA). In this study, results showed strong correlations between methylation levels of MGMT and DGKI of tDNA and cfcDNA in a cohort of 18 GBM patients (Supplementary Fig. 5). Importantly, no correlation was observed between MGMT and DGKI methylation levels in cfcDNA and those in buffy coat leukocyte DNA. This suggests that cfcDNA-based methylation measurements predominantly reflect tumor-derived DNA rather than leukocyte DNA, which may constitute up to 76% of the total cfcDNA [24] (Supplementary Fig. 5). Moreover, DGKI methylation levels in cfcDNA increased in three of the five patients analyzed (Fig. 4D), while MGMT methylation levels remained unchanged across all five (Fig. 4D). Together, these analyses indicate that TMZ treatment can alter DGKI methylation. Notably, three patients shifted from a favorable (MGMTM/DGKIU) to an unfavorable (MGMTM/DGKIM) methylation profile associated with reduced response to standard GB therapy (Fig. 4D).
Finally, A172 cells were co-treated with EXDDNMT1/ELK1 and TMZ for 8 weeks to assess whether this approach could prevent the emergence of TMZ resistance. Resistance was evaluated by measuring the IC₅₀ of cells that survived the co-treatment. These data show that TMZ + EXDDNMT1/ELK1 co-treatment prevents the emergence of TMZ-resistant A172 cells after 8 weeks of exposure, since the IC50 of these cells is 37 ± 6 μM, whereas it is 399 ± 36 μM when A172 cells are treated with TMZ alone (Fig. 4E).
Comments (0)