Cell lines and culture
THP-1-DualTM a dual reporter human monocyte cell line (InvivoGen, San Diego, CA, USA) was obtained for use in reporter assays, microscopy, and flow cytometry. This suspension cell line stably expresses a secreted alkaline phosphatase (SEAP) reporter inducible by NF-κB and a secreted lucia luciferase reporter inducible by IRF pathway activation. Knockouts for MyD88, TRIF, and TLR4 in the dual-reporter background were also obtained from InvivoGen. THP-1 cells were grown in RPMI 1640 with 2 mM L-glutamine (Cytiva, Marlborough, MA, USA) supplemented with 25 mM HEPES (Thermo Fisher, Waltham, MA, USA), 10% heat-inactivated FBS, and Pen-Strep (100 U/ml–100 µg/ml). Cells were maintained at 37 °C with 5% CO2. Selection antibiotics, Blasticidin (10 µg/ml) and Zeocin (100 µg/ml) (InvivoGen, San Diego, CA, USA), were applied every other passage per manufacturer recommendation.
A549-DualTM a dual reporter human respiratory epithelial cell line (InvivoGen, San Diego, CA, USA) was obtained for use in reporter assays. These cells were grown in DMEM with 2mM L-glutamine (Thermo Fisher, Waltham, MA, USA) supplemented with 10% heat-inactivated FBS and Pen-Strep (100 U/ml-100 µg/ml). Cells were maintained at 37 °C with 5% CO2.
mRNA preparation and lipid nanoparticle formulation
The diproline-modified SARS-CoV-2 Omicron variant spike glycoprotein (S2P) was codon optimized and cloned into an in vitro transcription template plasmid containing a T7 promoter, 5’ and 3’ UTR regions, and a 100 nucleotide poly(A) tail. mRNA was synthesized and co-transcriptionally capped using the Megascript Transcription kit (Thermo Fisher, Cat# AMB 1334) and the CleanCapTM dinucleotide system (Trilink Biotechnologies), precipitated, and purified using a modified cellulose-based chromatography method. Length and mRNA integrity were assessed using the Agilent Bioanalyzer 2100 system. Removal of double stranded RNA (dsRNA) contaminants was confirmed using dot blot, and endotoxin levels were measured using the Genscript ToxiSensor chromogenic assay (< 0.05 EU/mL). The S2P mRNA was stored frozen (1 mg/mL) at −20 °C in nuclease and pyrogen free water until use. mRNA was encapsulated into lipid nanoparticles as previously described (51). LNP lipid components were mixed in an ethanolic solution, and rapidly mixed with an aqueous phase containing the mRNA, dialyzed, and concentrated to 1 mg/mL, and stored at −80 °C until used (Supplementary Table 1, Supplementary Fig. 1).
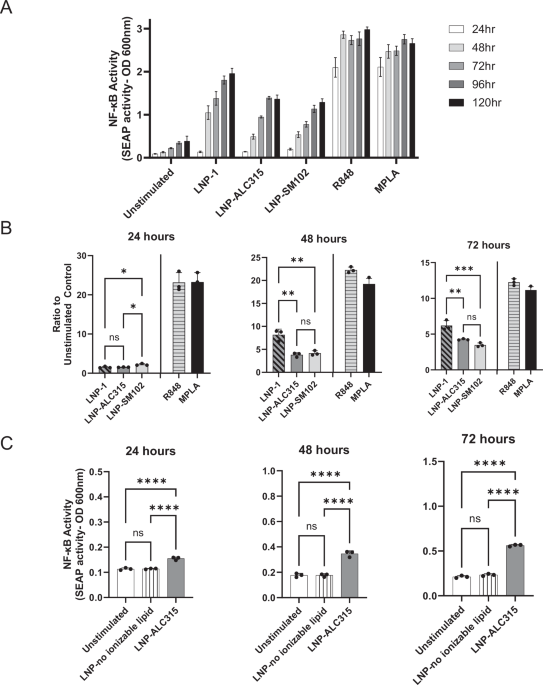
LNP-ALC315, following the BNT162b2 formulation, was comprised of the ALC-0315 ionizable lipid, cholesterol, DSPC, and the ALC-0159 PEG-lipid. LNP-SM102, following the mRNA-1273 formulation, was comprised of the SM-102 ionizable lipid, cholesterol, DSPC, and PEG-DMG PEG-lipid. A third LNP formulation, designated as LNP-1, is proprietary to Acuitas Therapeutics. The proprietary lipid and LNP composition are described in US patent US10,221,127. LNP-1 is comprised of the a proprietary ionizable lipid, DSPC, cholesterol, and a PEG-lipid. LNP-1 was also tagged with the DiO fluorescent marker and designated as DiO-LNP. The fourth formulation made was an LNP, designated as LNP-no ionizable lipid, containing only cholesterol, DSPC, and the ALC-0159 PEG-lipid. LNPs were characterized for their hydrodynamic size, and polydispersity index (PDI), using dynamic light scattering (DLS), and the encapsulation efficiency and concentration measured using the RiboGreen RNA assay (Invitrogen, Cat# R11490). The empty LNP-ALC315 used was 54.07 nm with a PDI of 0.28 and zeta potential of -4.447. The empty LNP-SM102 used was 51.590 nm with a PDI of 0.1605 and zeta potential of -4.417. The LNP without the ionizable lipid was larger at 80.26 nm with a PDI of 0.05 and zeta potential of -8.548.
mRNA-LNPs were made with LNP-1 and LNP-ALC315, designated as mRNA-LNP-1 and mRNA-LNP-ALC315. The mRNA-LNP-ALC315 had an encapsulation efficiency of 98.17%, 66.4 nm diameter, 0.1090 PDI, and zeta potential of −11.55. LNP concentrations are given as the mRNA equivalent to enable comparison with the mRNA-LNP as the concentration of mRNA.
Reporter assays
For both NF-κB and IRF reporter assays using the THP-1 cell line, cells were resuspended in RPMI 1640 with 2mM L-glutamine (Cytiva, Marlborough, MA, USA), 10% heat-inactivated FBS, and Pen-Strep (100 U/ml–100 µg/ml) at a concentration of 1 × 106 cells/ml and 1 ml aliquoted per well of a 24-well plate. Reagents used individually to treat THP-1 cells included the LNP-1, LNP-ALC315, LNP-SM102, LNP-no ionizable lipid, mRNA-LNP-1, mRNA-LNP-ALC315, VacciGrade R848 (InvivoGen, San Diego, CA, USA), synthetic MPLA (InvivoGen, San Diego, CA, USA), IFNɑ (Miltenyi, Gaithersburg, MD, USA), LPS (Sigma-Aldrich, St. Louis, MO, USA), and TNFɑ (Thermo Fisher, Waltham, MA, USA). Empty LNPs and mRNA-LNPs were generously provided by the Alameh lab and University of Pennsylvania synthesized as described above18.
For analysis of endocytosis, cells were either left untreated or treated with the endocytosis inhibitors dynasore (50 μM, Fisher Scientific, Hampton, NH, USA), cytochalasin D (500 μM, Fisher Scientific, Hampton, NH, USA), chloroquine (50 μM, Fisher Scientific, Hampton, NH, USA) or methyl-β cyclodextrin (250 μM, Thermo Fisher, Waltham, MA, USA). THP-1s were treated two hours before addition of stimuli and every 24 h afterward. After thorough resuspension of reagent with THP-1, 200 µL of supernatant was removed at the indicated time points for measurement of reporter molecules. Cell counts were taken prior to removal of supernatant on each day to ensure similar growth curves. For the NF-κB reporter assay, 20 µL of supernatant was mixed with 180 µL of Quanti-Blue Solution (InvivoGen, San Diego, CA, USA) in a 96-well plate and incubated at 37 °C with 5% CO2 for 24 h. The absorbance at 600 nm was read using the Promega GlowMax plate reader (Promega, Madison, WI, USA). For the IRF reporter assay, 20 µL of supernatant was mixed with 50 µL of Quanti-Luc Solution (InvivoGen, San Diego, CA, USA) and the luminescence was immediately read at 0.1 s integration using the Promega GlowMax plate reader. Biological and technical replicates were performed for each condition in each assay.
For both NF-κB and IRF reporter assays using the A549 cell line, 6.25 × 103 cells were seeded in each well of a 48 well plate with a final volume of 200 µL and incubated at 37 °C with 5% CO2 for 24 h. Supernatant was replaced with fresh media and stimulation was added. Cells were incubated for the designated time period and supernatant was removed for reporter assay as described above.
Fluorescent microscopy
Cells were incubated with or without 1 µg/ml DiO-tagged LNP (DiO-LNP) at a concentration of 5 × 105 cells/ml at 37 °C with 5% CO2 in 24 well plate. DiO-LNP was generously provided by the Alameh lab and the University of Pennsylvania synthesized as described above18. After incubation, cells were pelleted, washed, and resuspended in PBS. Cells were allowed to settle onto a coverslip at the bottom of 6-well plates for 30 min. Supernatant was gently removed, and cells were fixed with a 4% paraformaldehyde solution for 15 min. After fixation, cells were washed with PBS for two minutes twice. For intracellular staining, cells were permeabilized with saponin during fixation, blocked with 3% bovine serum albumin (BSA) (Miltenyi, Gaithersburg, MD, USA) for 1 h, and stained with anti-TLR4 antibody (anti-CD284) (Cat. No. 14-9917-82) (Thermo Fisher, Waltham, MA, USA) and anti-Rab5a antibody (Cat. No. 11947-1-AP) (Thermo Fisher, Waltham, MA, USA) for 1 h. Cells were subsequently washed with additional BSA and stained with secondary antibodies; anti-mouse IgG Alexa Fluor555 (Cat No. 4409S) (Cell Signaling Technology, Danvers, MA, USA) and anti-rabbit IgG Alexa Fluor647 (Cat No. 4414S) (Cell Signaling Technology, Danvers, MA, USA) for 1 h. Then, cells were stained with 1 µg/ml DAPI (BioLegend, San Diego, CA, USA) for 10 min in the dark and subsequently washed. Coverslips were mounted using ProLong Gold antifade reagent (Invitrogen, Waltham, MA, USA) and imaged on the Zeiss700 confocal microscope (Zeiss, Jena, Germany) using the ZEN 2.3 SP1 software.
Flow cytometry
THP-1 cells at a concentration of 1 × 106 cells/ml were incubated with DiO-LNP (1 µg/ml), LNP-1 (1 µg/ml), LNP-ALC315 (1 µg/ml), LNP-SM102 (1 µg/ml), mRNA-LNP-1 (1 µg/ml), mRNA-LNP-ALC315 (1 µg/ml), VacciGrade R848 (1 µg/ml) (InvivoGen, San Diego, CA, USA), or media alone at 37 °C with 5% CO2. For surface marker investigation, cells were washed with PBS and stained with Human TruStain FcX (BioLegend, San Diego, CA, USA) at 4 °C for 5 min to prevent nonspecific binding followed by surface staining with antibodies listed in Supplementary Table 2 at 4 °C for 20 min. Cells were fixed with a 4% paraformaldehyde solution for 15 min. For assessing kinetics of DiO-LNP uptake, cells were immediately washed and fixed. Cells were run on the Cytek Aurora Spectral Flow Cytometer and analyzed using FlowJo v10.
Sequencing
THP-1 cells at a concentration of 1 × 106 cells/ml were incubated with or without LNP-1 at 1 µg/ml for 24 h at 37 °C prior to RNA extraction with Qiagen RNeasy Plus Mini Kit (Qiagen, Hilden, Germany). Each condition was set up in quadruplicate with 1 million cells per sample. RNA was quantified using Qubit RNA Broad Range Assay Kit (Thermo Fisher, Waltham, MA, USA). For all samples, a total RNA amount of >100 ng was used as input for library preparation using the TruSeq Stranded total RNA Library Preparation Kit (Illumina, San Diego, CA, USA) using IDT for Illumina – TruSeq RNA UD Indexes. Sequencing libraries were quantified by real-time PCR using the KAPA Library Quantification Complete kit (Roche, Basel, Switzerland) and assessed for size distribution and absence of free adapters and adapter dimers on a Fragment Analyzer. Sequencing libraries were pooled and quantified by real-time PCR as above, clustered and sequenced on an Illumina NovaSeq 6000 using a Reagent Kit v1.5 (200 cycles) with run parameters generating paired-end reads at 100 bp length.
Sequencing analysis
Fastq files obtained from the sequencing run were initially processed through the nf-core rnaseq pipeline via NextFlow using recommended pipeline settings75,76. The pipeline includes FASTQC, Trim Galore, SortMeRNA, STAR, featureCounts, StringTie, Salmon, Qualimap, dupRadar, Preseq, and RSeQC. Reads were aligned to the GRCh37 reference genome. Subsequent analysis was performed with R 4.3.2 using Jupyter Notebooks and the Tidyverse (v2.0.0) package. We used DESeq2 (v1.42.0) to identify differentially expressed genes (DEGs) between unstimulated and LNP-1-stimulated samples with 4 samples in each group. The Benjamini-Hochberg procedure was used for multiple testing correction. Additionally, we implemented a false discovery rate of 0.01 and a cutoff of a logfold change above 1 or below -1. Then, we performed logfold change shrinkage using apeglm (v1.24.0)77 and matched gene IDs to gene symbols to create a table of DEGs. The table of DEGs was run through Human MSigDB Collections (v7.5.1): hallmark gene sets (H), KEGG (C2: curated gene sets), and the GO (C5: ontology gene sets - GO: Gene Ontology gene sets). Gene Set Enrichment Analysis (GSEA) was performed using the Bioconducter clusterProfiler (v4.8.3) package.
Statistical analysis
Statistical analyses were performed using GraphPad PRISM (v10, GraphPad Software Inc., Boston, MA, USA) using a one-way ANOVA followed by Dunnet’s test for multiple comparisons to a single control or by Tukey’s test for multiple comparisons between all groups. Results are expressed as means with standard error of the mean (SEM). Values of P < 0.05 were considered statistically significant. For transcriptomics data, the Benjamini-Hochberg procedure was implemented during determination of DEGs and pathway enrichment analysis to correct for multiple comparisons. The adjusted p-values were reported for these results. Significance for GSEA was determined by comparing the enrichment score to a null distribution through permutation testing.

Comments (0)