Mice
Wild-type (WT) C57BL/6 (B6) and BALB/c mice (6–8 week old, female) were purchased from Orient Bio (Seongnam, South Korea). OT-I (C57BL/6-Tg(TcraTcrb)1100Mjb/J) and OT-II (B6.Cg-Tg(TcraTcrb)425Cbn/J) mice were purchased from Jackson Laboratories (ME, USA). Mice were maintained under specific pathogen-free conditions. Anesthesia and euthanasia were performed following the approved IACUC protocols. For subcutaneous injection, submandibular (facial) bleeding, and intranasal virus challenge, mice were anesthetized with isoflurane inhalation in an anesthesia jar. For tissue sampling and terminal bleeding (via cardiac puncture), mice were euthanized with exposure to CO2. The animal experiments were approved by the Institutional Animal Care and Use Committee of Institut Pasteur Korea (IACUC approval no. IPK-20012-C2, IPK-21003-D2, and IPK-23005).
Immunization
Mice were vaccinated with single or double (prime-boost) doses of antigens and additives as described below. Immunization was performed subcutaneously at the base of the tail, in less than 150 μL of inoculum. For the immunization, we used following materials: endotoxin-free ovalbumin (Ova) (10 μg/mouse for primary and 50 μg/mouse for secondary immunization, Worthington, OH, USA), influenza A/Puerto Rico/8/1934(H1N1)(PR8) hemagglutinin (HA) protein (0.1 or 0.5 μg/mouse, SinoBiological, China), AV (50 μL/mouse, Invivogen, CA, USA), uricase (10 unit/mouse, Sigma-Aldrich, MO, USA), febuxostat (100 μg/mouse, Sigma-Aldrich), and monosodium urate (MSU, 500 μg/mouse, Invivogen). AV was used as a preclinical grade SE adjuvant with a formulation similar to that of MF59.
Viral challenge experiments
Vaccinated mice were intranasally infected with a highly lethal dose of the PR8 virus (400 PFU, 20\(\times\) LD50 for a single dose and 2000 PFU, 100\(\times\) LD50 for a double dose vaccination model), using 20 μL of inoculum per mouse, administered by placing droplets on the nostrils and allowing inhalation of the solution. After infection, the mouse body weight was monitored daily for 14 days, and the mice were euthanized at the humane endpoint. The humane endpoint was determined based on a scoring system measuring body weight loss, physical appearance, and behaviors. Since the infected mice with > 25% weight loss typically reached score of endpoint by revealing anorexia, progressive dehydration, and reduced mobility, this was the final criterion for scoring death and performing euthanasia.
DAMP assays
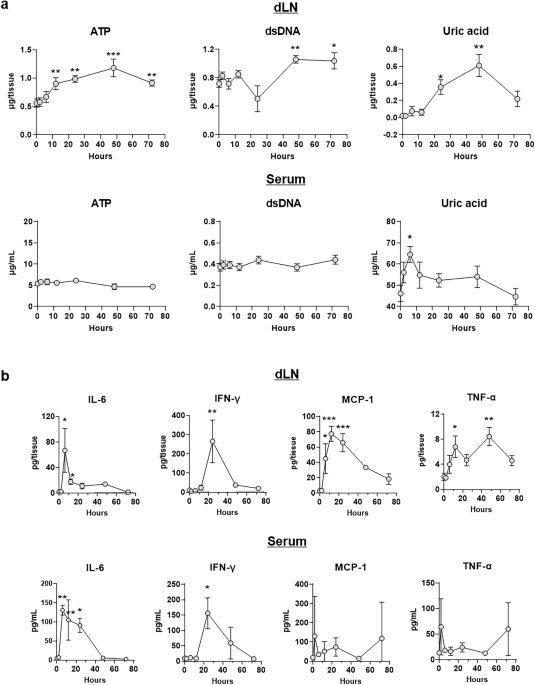
Serum and inguinal LN samples were collected at the indicated time points. For serum separation, blood samples were clotted for 1 h at 4 °C and centrifuged at 10,000\(\times\) g at 4 °C for 10 min. To prepare LN fluids without rupturing cells, two inguinal LNs from each mouse were dissociated into a single-cell suspension by grinding them on a 70 μm cell strainer in 300 μl of PBS. Supernatants were collected by centrifugation at 300\(\times\) g for 10 min at 4 °C. DAMP assays were performed using UA and ATP assay kits (Abcam, Cambridge, UK), and a Quanti-iT PicoGreen dsDNA assay kit (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s protocol.
Flow cytometry
Flow cytometry was performed to detect cell surface proteins and intracellular cytokines in single-cell suspensions of various tissues and immune cells. Single-cell suspensions were prepared by dissociating the tissues through a 70 μm cell strainer. For the preparation of innate immune cells, tissues were treated with collagenase type IV (Worthington, 400 U/tissue) at 37 °C (30 min for the spleen, 15 min for LNs) prior to passing through the cell strainer. The following antibodies were purchased from BD Biosciences (NJ, USA) for detection of each molecules: CD8 (53-6.7), CD4 (RM4-5), CD95 (Jo2), CD279 (PD-1) (J43), CD185 (CXCR5) (2G8), CD80 (16-10A1), NK1.1 (PK136), CD317 (PDCA-1) (927), CD11b (M1/70), Siglec-F (E50-2440), and CD69 (H1.2F3). The following antibodies were purchased from Biolegend for detection of each molecules: CD62L (MEL-14), CD19 (6D5), GL7 (GL7), CD3 (17A2), Ly6G (1A8), Ly6C (HK1.4), CD86 (GL1), CD11c (N418), F4/80 (BM8), MHC II (I-A/I-E) (M5/114.15.2), IL-2 (JES6-5H4), TNF (MP6-XT22), IFN-γ (XMG1.2), and CD185 (CXCR5) (L138D7). Staining was performed with a mouse Fc block (BD Biosciences). The H2Kb-SIINFEKL reagent was provided by the NIH Tetramer Core Facility (GA, USA). To assess cell viability, we used the following viability kits prior to staining: Zombie Aqua or Zombie NIR (BioLegend). For intracellular cytokine staining, the Cytofix/Cytoperm solution kit (BD Biosciences) was used according to the manufacturer’s protocol. Stained cells were analyzed using CytoFLEX LX (Beckman Coulter, Brea, CA, USA), and the data were analyzed using FlowJo software (BD Biosciences).
Bead-based cytokine array and cytokine ELISA
Mouse inflammatory cytokine levels in LN fluid and serum were analyzed using bead-based cytokine assays (LEGENDplex mouse inflammation panel, BioLegend), according to the manufacturer’s instructions. LN fluids were used for the cytokine array without dilution, and serum samples were 1:1 diluted with the assay buffer. All samples were analyzed using CytoFLEX LX (Beckman Coulter). Mouse IL-18 and CXCL10 levels were determined using DuoSet ELISA kits (R&D systems).
Antibody ELISA
Antigen-specific immunoglobulin (Ig) levels were determined by ELISA, as described previously40 using 96-well EIA/RIA high-binding plate (Corning, NY, USA) coated with Ova (2 μg/mL in PBS, Worthington) or HA (2 μg/mL in PBS, SinoBiological). We used the following antibodies conjugated with horseradish peroxidase (HRP): goat anti-mouse IgM, goat anti-mouse IgG, goat anti-mouse IgG1, goat anti-mouse IgG2b, and goat anti-mouse IgG2c (all from Southern Biotech, AL, USA). HRP activity was measured using the BD OptEIA Reagent Set (BD Biosciences) and stopped using 2 N H2SO4.
Plaque reduction neutralization test (PRNT)
Mouse sera were serially diluted in infection media (DMEM supplemented with 0.3% BSA and 2 μg/mL TPCK-treated trypsin). Each diluted sample was mixed with an equal volume of virus suspension (100 pfu/well) and incubated at room temperature for 1 h. MDCK cells were cultured to confluence in 12-well cell culture plates 24–36 h before the experiment, washed with PBS, and infected with each serum-virus mixtures at 37 °C for 1 h. After incubation, the mixtures were removed, and the cells were overlaid with agarose media (0.5% agarose, DMEM with 0.3% BSA and 2 μg/mL TPCK-treated trypsin at final concentration). The plates were incubated at 37 °C with 5% CO2 for 48 h. Following incubation, the cells were fixed with 4% paraformaldehyde at room temperature for at least 15 min before removing the agarose overlay. The fixed cells were then stained with 0.1% crystal violet, and washed with water. Plaques were counted after the plates were dried. PRNT50 titers were calculated as the reciprocal of the lowest serum dilution at which 50% of the virus was neutralized compared to the infection control (with no serum).
Intracellular cytokine staining
Cells from the LNs and spleens were stimulated with Ova peptides in media at 37 °C for 5 h in the presence of the protein transport inhibitors GolgiStop and GolgiPlug (BD Biosciences), in 96-well cell culture plates (1 × 106 cells/well). We used a mixture of Ova MHC class I peptide epitope (OVA 257–264, Invivogen) and the Ova MHC class II epitope (OVA 323–339, Invivogen). The cells were subjected to cell surface and intracellular cytokine staining.
Generation of bone marrow-derived dendritic cells (BMDCs) and macrophages (BMMs)
BMDCs and BMMs were prepared by culturing B6 female mouse bone marrow cells in RPMI-1640 (Welgene, South Korea) supplemented with 10% heat-inactivated fetal bovine serum (Gibco), 10 mM HEPES (Welgene), 100 U/mL penicillin and 100 μg/mL streptomycin (Welgene), and 50 μM β-mercaptomethanol (Sigma-Aldrich), supplemented with 2 mM L-glutamine (Welgene) for BMDCs and 2 mM sodium pyruvate (Welgene) for BMMs. Cell cultures were supplemented with 20 ng/mL GM-CSF for BMDC differentiation or 20 ng/mL M-CSF for BMM differentiation (Miltenyi Biotec, Germany). BMDCs were harvested by collecting non-adherent cells on day 10. BMMs were harvested on day 6 by collecting adherent cells using trypsinization.
DC-T cell proliferation assay
The antigen presentation capacity of the DCs was evaluated by T cell proliferation after DC-T cell co-culture. BMDCs were stimulated with Ova (80 μg/mL), Ova + MSU (100 μg/mL) or Ova + MSU (300 μg/mL) for 20 h in 96-well round-bottom cell culture plates (SPL Life Sciences, Pocheon, South Korea). For DC recovery from the MSU culture, we isolated CD11c+ cells using anti-CD11c magnetic beads (Mojosort nanobeads, BioLegend) and a magnet (Easysep magnet, STEMCELL Technology, Vancouver, Canada). To isolate OT-I or OT-II cells, we obtained the spleen and LNs from each mouse and enriched the naïve OT-I or OT-II cells by negative selection using a mouse pan-naïve T cell isolation kit (EasySep, STEMCELL Technology). To track the proliferation of T cells, we stained the isolated T cells with 0.5 μM CFSE (CellTrace cell proliferation kit, Invitrogen, MA, USA) at 37 °C for 5 min, followed by quenching on ice for 5 min. A mixture of DCs and CFSE-labeled T cells was incubated for 3 days, and CFSE dilutions were measured by flow cytometry. The ratios of cell number between the DCs and T cells were 1:1, 1:5, and 1:10. T-cell CFSE levels were measured on days 0 and 3.
Stimulation of immune cells with MSU
BMDCs and BMMs were cultured from WT B6 mouse bone marrow cells, as described above. Splenic B and T cells were isolated using an EaspSep mouse B cell isolation kit and a pan-naïve T cell isolation kit (STEMCELL Technology). BMDCs (5 × 105 cells/well) in 24-well non-TC cell culture plates (Nunc, Roskilde, Denmark), BMMs (5 × 105 cells/well), splenic B (5 × 105 cells/well), and T cells (2 × 105 cells/well) in 24-well cell culture plates (SPL Life Sciences) were stimulated with MSU as indicated, either with or without 100 ng/mL LPS priming. Activation marker expression levels were analyzed after stimulation with 100 or 300 μg/mL MSU for 24 h, by flow cytometry. RNA sequencing was performed after stimulation with 300 μg/mL MSU for 3 h.
RNA sequencing and data analysis
Total RNA was extracted using the TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. The RNA amount was quantified using nanodrop (Thermo Fisher Scientific), and ≥2 μg RNA per sample was used for RNA sequencing.
RNA sequencing was performed by ROKIT Genomics (South Korea). For the analysis of the bulk RNA sequencing data, we used the mouse genome (mm10_NCBI_108)41 along with gene annotations from mm10.refGene.gtf42. We employed Hisat2 v2.1.043 for mapping and alignment, and featureCounts v2.0.3 for counting44. After each step, we conducted quality control checks using FastQC 0.11.9 and MultiQC v1.1245,46. To identify differentially expressed genes (DEGs), raw counts were obtained from the sequencing data. Genes with zero counts across all of the samples were removed. We then searched for unusual values or outliers using Cook’s distance from the DESeq2 1.40.2 package47 and the Molecular Degree Perturbation (MDP) score from the mdp 1.24.0 package in R48. For the actual differential expression analysis, we used the default parameters of the DESeq2 package47. We considered genes with padj < 0.01 and │log2FC│ > 1 as statistically significant DEGs.
For functional enrichment analysis, we selected DEGs using a statistical cutoff of a padj < 0.05 and │log2FC│ > 1. We ranked these DEGs by log2FC and conducted a pre-ranked gene set enrichment analysis (GSEA) using the fgsea 1.28.0 package49. Comprehensive annotation datasets were used, including “BioCarta 2016,” “BioPlanet 2019,” “GO Biological Process 2021,” and others50,51,52,53,54,55,56,57. To ensure robust and relevant pathways, we excluded those with sizes < 15 or > 500 during enrichment analysis. We considered pathways with padj < 0.05 as statistically significant. The analyses were performed using the fgsea and hypeR 2.0.1 packages in R software49,58.
Statistical analysis
All statistical analyses were performed using Prism 9 software (GraphPad Software, CA, USA). Normal distribution was evaluated using the Shapiro-Wilk test before determining statistical significance (normal distribution if p > 0.01). Equality of variance was also examined with the Brown-Forsythe test for more than three groups and with the F-test for comparison of two groups. For comparison of the three groups, statistical significance was determined by one-way ANOVA, Welch ANOVA, or the Kruskal-Wallis test, depending on the normal distribution and variance equality, followed by multiple comparison tests using the two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli. For comparison of data between the two groups (AV vs. AV+febuxostat), we used the Mann-Whitney U test, unpaired t-test, or Welch’s t-test, depending on normal distribution and variance equality. Survival data were analyzed using the log-rank test. Statistical significance is indicated by P values (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).

Comments (0)